
CRISPR/Cas9介导烟草多基因编辑体系的应用
2019-09-04 12:20:04谢小东高军平李泽锋张剑锋魏攀罗朝鹏王晨武明珠翟妞杨军
中国烟草学报 2019年4期
谢小东,高军平,李泽锋,张剑锋,魏攀,罗朝鹏,王晨,武明珠,翟妞,杨军
1 中国烟草总公司郑州烟草研究院,国家烟草基因研究中心,郑州高新技术产业开发区枫杨街2号 450001;
2 湖南中烟工业有限责任公司技术研发中心,长沙市劳动中路386号 410008
基因编辑技术可以精准地对基因组中的靶位点进行缺失、敲入以及核苷酸修正等操作。CRISPR/Cas9(clustered regularly interspaced short palindromic repeats/CRISPR-associated nuclease 9,Cas9)基因组编辑技术问世以来,弥补了第一代的锌指核酸酶(zinc finger nucleases,ZFN)和第二代的转录激活效应子核酸酶(transcription activator-like effector nucleases,TALEN)基因编辑技术的诸多不足。因CRISPR/Cas9系统靶向编辑基因的高效性、特异性和设计的便捷性,已成为当前最主流、最热门的基因组编辑技术,在大量物种中得到了广泛应用。
CRISPR/Cas9是细菌和古细菌在长期演化过程中形成的一种适应性免疫防御系统,可抵抗病毒或外源性DNA的侵染。CRISPR/Cas9编辑技术是利用导向RNA识别靶位点,并引导Cas9蛋白切割靶序列[1-2]。CRISPR/Cas系统有3种类型(Type Ⅰ、Type Ⅱ和Type Ⅲ)。现在广泛用于基因编辑技术的CRISPR/Cas系统是根据Type II型系统改造而来的。Type Ⅱ型系统由Cas9核酸酶、pre-crRNA和反式激活crRNA组成。在前导序列的调控下,CRISPR的间隔序列被转录成为pre-crRNA。借助tracrRNA,RNase III再将pre-crRNA加工为成熟的crRNA,并形成具有切割活性的tracrRNA-crRNA-Cas9复合体。随着对CRISPR/Cas9技术的不断优化,crRNA和tracrRNA被融合成为一条单链引导RNA(singleguide RNA,sgRNA)。sgRNA的表达通常用III型RNA聚合酶启动子转录,PAM(protospacer adjacent motif)序列决定CRISPR/Cas9系统切割的位点[3]。随着基因编辑技术的迅速发展,科学家开发了不同CRISPR/Cas9系统,不断在提升CRISPR/Cas9基因编辑技术应用价值。
2013年,科学家首次利用CRISPR/Cas9系统实现了对拟南芥[4]、水稻和小麦基因组的定点编辑[5],证明了该技术在植物生物学研究领域具有极高的应用价值。随后CRISPR/Cas9系统被广泛应用于高粱[6]、玉米[7]、甜橙[8]、番茄[9,10]、大豆[11]、大麦[12]、甘蓝[12]、棉花[13]、甘蔗[14]等作物中,在性状改良、基因调控、抗性育种以及高通量突变体文库创制等方面得到了广泛应用[15]。高军平等利用CRISPR/Cas9系统分别对烟草NtPDS和NtPDR6基因实现了定点突变[16],首次在普通烟草中建立了CRISPR/Cas9技术体系。本课题组利用CRISPR/Cas9技术在烟草中鉴定了一个腋芽生长相关基因NtPIN4,将基因突变后烟草腋芽数目比野生型明显增多,且突变类型和表型均能够稳定遗传至下一代[17]。潘洪杏等利用CRISPR/Cas9技术对烟草马铃薯Y病毒(Potato Y virus,PVY)基因eIF4E进行了定向编辑[18]。姚恒等利用CRISPR/Cas9技术对烟草茉莉酸信号关键转录因子基因NtabMYC2进行了编辑,获得了不同敲除类型的突变体[19]。
随着基因编辑技术的快速发展,在许多物种中同时编辑多个基因的技术逐步成熟。在杨树毛白杨中,通过构建多靶点编辑载体系统,实现了同时敲除杨树基因组上两个同源的PDS编码基因(PtPDS1和PtPDS2)[20]。在水稻和拟南芥中,构建的多基因敲除体系能够成功突变拟南芥和水稻基因家族的多个基因、以及单个基因的多个靶点,在转化体第一代就获得有表型的突变体[21]。利用CRISPR/Cas9系统在水稻育种中间材料中同时编辑TMS5、Pi21和Xa13基因,成功将具有优良性状的育种中间材料快速开发成优质多抗的两系不育系[22]。通过构建多基因敲除体系,同时对编码拟南芥核糖体大亚基AtRPL10亚家族的三个同源基因AtRPL10A,AtRPL10B及AtRPL10C进行了编辑,转基因植株的三个同源基因均存在基因突变[23]。
烟草是异源四倍体农作物,多基因编辑体系的建立与应用,对加快烟草基因功能的研究和遗传改良的有着重要的意义。在烟草中,针对多个基因、尤其是不同性状相关基因的同时编辑的研究尚未有报道。为此,基于烟草中已有的CRISPR/Cas9系统,本文选取以下5个靶基因开展CRISPR/Cas9介导多基因编辑研究。eIF4E是转录起始因子(eukaryotic translation initiation factors 4E),PVY病毒通过自身的基因组连接蛋白(viral genome-linked protein,VPg)与植物的特定eIF4E基因家族成员相互作用,从而启动病毒在植物体内的翻译与增殖。eIF4E基因突变会导致其表达的蛋白结构改变或缺失,使病毒无法利用eIF4E蛋白侵入植株[24]。本生烟、辣椒以及普通烟草中突变或者下调eIF4E基因,均能使植株对PVY产生一定的抗性[25,26]。TOM1和TOM3是烟草花叶病毒(Tobacco mosaic virus,TMV)的抗性相关基因,研究发现TOM3蛋白与TMV病毒特异的结合,进而将病毒复制复合体整合到质膜之上,保证病毒的复制[27]。Sunil Kumar等通过构建TOM1和TOM3的干扰载体,进而下调烟草中的这两个基因,发现转基因植株对TMV的抗性明显增强[28]。在本氏烟中克隆了拟南芥同源基因NbTOM1,分析证明病毒诱导NbTOM1沉默后,几乎完全遏制了本氏烟中TMV的复制,同时干扰沙姆逊烟草的TOM1和TOM3基因后,发现转基因烟草能够有效地抑制TMV的生长和扩散[29]。课题组前期克隆了烟草植物螯合肽基因NtPCS1,利用RNAi技术降低NtPCS1基因的表达量之后,烟草叶片中的镉含量显著降低。利用TILLING技术将镉转运基因NtHMA2的突变后,在镉胁迫下,与对照相比突变体烟叶中的镉含量降低36.84%[30]。本文通过对上述5个烟草不同性状的基因开展CRISPR/Cas9多基因编辑的探索研究,旨在为烟草多个(性状)基因的功能研究和遗传改良提供技术依据。
1 材料与方法:
1.1 材料
普通栽培烟草K326无菌苗培养于国家烟草基因研究中心的人工气候室:将K326种子放入1.5 mL离心管中,加入1 mL 15% 次氯酸钠溶液消毒15 min,无菌水清洗5~6次,用牙签播种于MS固体培养基中。MS固体培养基成分为:4.4 g/L MS(Murashinge & Skoog Basal Mediun) 培养基、30 g/L蔗糖、2.5 g/L植物凝胶。培养条件为相对湿度60%,光照16 h/28 ℃,黑暗8 h/25 ℃。
1.2 方法
1.2.1 sgRNA设计及载体构建
利用烟草基因组序列信息,通过在线工具CRISPR MultiTargeter(http://www.multicrispr.net/index.html),设计了如下的编辑位点(表1)。将各靶位点序列分别接上U26启动子和终止后依次串联,并在串联的5'端和3'引入酶切Bsa I位点后合成序列,然后利用BsaI酶切,并与预先用BsaI酶切过的CRISPR/Cas9载体连接。连接体系:酶切载体 3 μL,退火产物 2 μL,Solution I 5 μL。16 ℃连接30 min,取连接产物转化 DH5α感受态细胞,37 ℃培养 12~16 h。用 U26-JC F:5'-TTAGGTTTACCCGCCAATA-3'和各靶序列下游引物对菌斑进行阳性克隆的PCR扩增、克隆和测序筛选。
1.2.2 烟草PCS1、eIF4E、TOM3、TOM1和HMA2基因敲除靶位点的cDNA克隆
以中国烟草基因组数据库中PCS1、eIF4E、TOM3、TOM1和HMA2基因的cDNA序列,设计可扩增PCS1、eIF4E、TOM3、TOM1和HMA2sgRNA序列的cDNA片段引物。PCS1-cl F:5'-GAAGACTCCAAGACTACCGGG-3',PCS1-cl R:5'-GCCAATTCGCAACTAACTTCA-3',eIF4E-cl F:5'-GTAGTCGACGATGGACCTGAA-3',eIF4E-cl R:5'-CATGAAATATGAAGCCAATCG-3',TOM3-cl F:5'-GGATGGGCTTAGACCTAGTTT-3',TOM3-cl R:5'-TTAGCGAATAGGGTGGTACTG-3',TOM1-cl F:5'-TGTTGTAAATGGAGTTCGTGC-3',TOM1-cl R:5'-CTCTTTTTGGAGGCAATTTTC-3',HAM-cl F:5'-GCATTAGCAACAGCTGACATT-3',HAM-cl R:GCGATGAGAATGAAAACCAC-3',以 K326品种的cDNA为模板进行PCR扩增反应。PCR反应体系(25 μL):基因上、下游引物(10 μM)各1 μL、DNA样品(100 ng/μL)1 μL、Taq酶12.5 μL,加 ddH2O补足至 25 μL。PCR 反应程序:94 ℃ 预变性5 min;94 ℃变性 30 s,58 ℃退火30 s,72 ℃ 延伸40 s,30个循环;72℃延伸10 min,4℃保温。待PCR结束后进行琼脂糖凝胶电泳,回收纯化PCR产物。用TOPO-TA PCR克隆试剂盒进行TA克隆,转化DH5α感受态细胞,37 ℃培养12~16 h,菌落PCR阳性单克隆送测序。
1.2.3 敲除载体转化农杆菌及烟草遗传转化
将测序成功的单菌落扩大培养,提取质粒,利用电转法将重组编辑载体转入农杆菌GV3101。具体的遗传转化方法见王姗姗 等[33]。
1.2.4 遗传转化株的获得及突变位点检测
待再生植株的根系生长良好后,移出组培瓶于基质中,置于培养条件为相对湿度60%,光照16 h/28 ℃、黑暗8 h/25 ℃人工气候培养箱中培养。采集适量烟株叶片,提取基因组DNA。具体步骤参照GeneAnswer GenePure新型植物基因组DNA快速提取试剂盒说明书。设计Cas9蛋白基因检测引物,Cas9-JC F:5'-CTCAACACAACATATACAAAACAAA-3',Cas9-JC R 5'-CTTTGGCCATCTCGTTTGA-3',以各植株DNA为模板进行PCR扩增,确认外源DNA片段是否已经插入到植物基因组中。PCR反应体系和程序同1.2.2。
将滚子从动件的滚子中心视为尖底从动件的尖底,则滚子从动件的凸轮机构即成为尖底从动件的凸轮机构,因此文中仅研究后者。
设计扩增包含靶位点的150bp-180bp DNA片段的引物,以检测目的基因是否成功被编辑。PCS1-JC F:5'-ATTGCCAGCAGTAACTTTGATGTT-3',PCS1-JC R:5'-GCCTAGTCCTAATTTTTTACTTGCC-3',eIF4EJC F:5'-ATTGGACAATGAGCTTTAGTAAGGG-3',eIF4E-JC R:5'-TTCATTTGCAGCATTCTTGGTC-3',TOM3-JC F:5'-ATGAAAGGTGGAGTAAT ACTTGTCT-3',TOM3-JC R:5'-TCAATCAT ATCCAGGTAAGAAAAGT-3',TOM1-JC F:5'-CTGTTCATTGGGAATTCGTAT TOM1-JC R:5'-GGGAGGAGAATGTAGTGAGGAA,HMA2-JC F:5'-AGCAGGTTATCCATTGGTTTG-3',HMA2-JC R:5'-TGACATCTTGGAACACACACC-3'。分别在以上各正向引物的5'端前加ggagtgagtacggtgtgc,反向引物5'前加gagttggatgctggatgg。以Cas9阳性烟株DNA为模板,进行PCR扩增。PCR反应体系和程序同1.2.2。待PCR结束后进行琼脂糖凝胶电泳,备用进行第二轮扩增。第二轮PCR 反应体系(20 μL):ddH2O 9.0 μl、Hi-TOM Mix 10 μl、第一轮 PCR 产物1.0 μl。94℃,预变性 2 min;94 ℃变性 30 s,58 ℃退火30 s,72 ℃ 1 kb/min,35个循环;72 ℃延伸5 min。PCR 结束后取3-5 μL 产物进行琼脂糖凝胶电泳检测。将扩增完毕的第二轮PCR 产物混合,取混合后的PCR 产物200 μL,进行琼脂糖凝胶电泳,切胶回收纯化。送北京诺禾致源生物信息科技有限公司进行高通量测序。
1.2.5 脱靶分析
以各基因靶位点序列为种子序列,分别在烟草基因组中数据库进行Blast比对,在全基因范围内寻找可能存在的脱靶位点。在候选脱靶位点序列位点上、下游设计引物扩增,通过测序的方法进行验证。
2 结果与分析
2.1 多基因敲除靶位点的sgRNA设计
利用中国烟草基因组数据库中PCS1、eIF4E、TOM3、TOM1和HMA2基因的cDNA序列,通过CRISPR MultiTargeter在线软件对PCS1、eIF4E、TOM3、TOM1和HMA2设计了 CRISPR/Cas9敲除的sgRNA寡聚核苷酸序列,再将设计的各sgRNA序列与基因组DNA进行了联配分析,筛选出高特异性sgRNA,并排除sgRNA序列夸两个外显子的可能性,获得如表1所示的候选的sgRNA。

表1 多基因敲除的sgRNA寡聚核苷酸序列Tab.1 The sgRNA oligonucleotide sequences of multiple genes
2.2 烟草PCS1、eIF4E、TOM3、TOM1和HMA2基因的sgRNA区域验证
为了排除基因组测序过程中造成的碱基误差,确保敲除靶位点序列的特异性和准确性,进一步对含有靶位点sgRNA序列的cDNA片段进行了测序验证。通过设计特异性扩增引物,以K326品种的cDNA为模板,分别扩增了PCS1、eIF4E、TOM3、TOM1和HMA2含有靶位点序列的cDNA片段,如图1所示,从K326中均扩增到预期大小的cDNA片段,并进行了纯化与克隆。测序分析显示,PCS1、eIF4E、TOM3、TOM1和HMA2cDNA片段中的sgRNA序列和PAM区与表1中所设计的sgRNA序列及PAM区完全一致,可以作为Cas9对相应基因敲除的sgRNA。

图1 烟草PCS1、eIF4E、TOM3、TOM1和HMA2 cDNA片段电泳检测与测序Fig.1 Electrophoresis detection and sequencing of partial cDNA sequence of tobacco PCS1,eIF4E,TOM3,TOM1 and HMA2
2.3 CRISPR/Cas9介导多基因敲除的sgRNA设计与构建
在每个sgRNA上下游分别引入拟南芥U-26启动子和终止序列,用于独立启动和终止sgRNA表达,再将各独立的sgRNA表达元件依次串联后,在5'端和3'引入酶切BsaI位点,获得如图2A所示的串联sgRNA片段。利用BsaI酶切的方法将该串联片段连接至pORE-Cas9植物表达载体上。分别以U26-JC F和PCS1、eIF4E、TOM3、TOM1和HMA2sgRNA的反向引物进行PCR扩增,通过凝胶电泳检测显示,5个sgRNA以及载体片段均被扩增到,并呈现出按序排列清晰条带(图2B),符合预期目标。这表明已经成功构建了针对PCS1、eIF4E、TOM3、TOM1和HMA2基因敲除的CRISPR/Cas9表达载体。
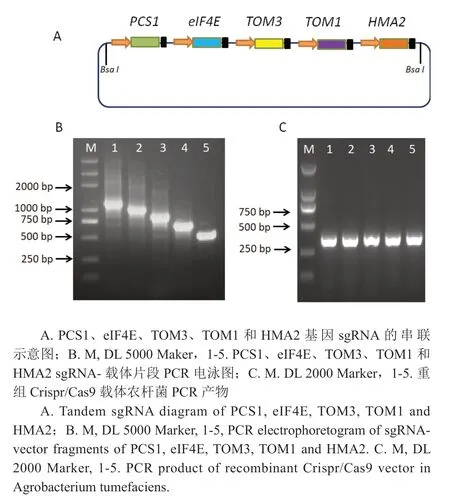
图2 多基因敲除的Crispr/Cas9载体构建Fig.2 Construction of Crispr/Cas9 vector for multi-gene knockout
提取重组CRISPR/Cas9载体质粒,采用电击法转入农杆菌GV3101,对经过卡那霉素和利福平筛选的单菌落进行菌落PCR验证,所用引物为U26-JC F和PCS1、eIF4E、TOM3、TOM1和HMA2sgRNA的反向序列。如图2C所示,得到预期300 bp左右大小的目的条带,表明重组CRISPR/Cas9载体质粒已经成功转入农杆菌GV3101菌株内。
2.4 T0代遗传转化植株的检测
利用农杆菌介导的叶盘法将重组CRISPR/Cas9载体质粒转化烟草K326,经过卡那霉素筛选,获得了17株T0代遗传转化阳性苗。待幼苗长至4~6片叶时,提取叶片基因组DNA,以检测编码Cas9蛋白基因的Cas9-JC F和Cas9-JC R引物进行PCR扩增。琼脂糖凝胶电泳结果显示,从13株烟草中扩增出预期500 bp左右的Cas9基因片段(图3),表明构建的重组CRISPR/Cas9基因敲除质粒已成功转入K326植株中。
2.5 烟草PCS1、eIF4E、TOM3、TOM1和HMA2突变靶位点的扩增与突变分析
设计特异性引物,以各阳性植株DNA为模板,扩增包含突变靶位点在内的150 bp-180 pb的PCR产物。第二轮PCR扩增以第一轮PCR产物为模板,利用Hi-Tom试剂盒进行扩增与加测序接头的PCR反应,其产物比第一轮长100 bp左右。第一轮和第二轮PCR扩增产物片段大小如图4所示,纯化回收后进行建库测序。
测序结果分析显示,PCS1基因在靶位点处发生了多种形式的突变,包括在不同位点处的1个、2个、4个、5个和10个碱基的删除和1个碱基的插入(图5A),大多数突变发生在PAM区域附近。Cas9对TOM3基因同样进行了成功编辑,分析显示在TOM3-sgRNA序列处主要发生了1个碱基和2个碱基的删除(图5 B)。另外,在TOM3-sgRNA位点处分别发现了1个碱基的替换和1个碱基的插入突变。在TOM1-sgRNA序列处有4个不同位点1个碱基的删除、一个位点2个碱基的删除以及2个不同位点T→C的碱基替换(图5C)。在eIF4E-sgRNA位点主要以碱基删除为主,包括4个不同位点1个碱基的删除、1个位点4个碱基和1个位点2个碱基的删除(图5D),这三种形式的删除突变均可导致eIF4E基因在烟草中的功能丧失。对HMA2基因的测序结果分析显示,HMA2-sgRNA位点处不仅有1个碱基的删除,还存在3个、6个和8个碱基的删除(图5E)。根据靶位点中碱基突变的reads与总测序reads的比值,评估了Cas9对靶位点处编辑效率。Cas9对PCS1、TOM3、TOM1、eIF4E、HMA2靶位点处的编辑效率分别约为13.1%、15.1%、6.1%、19.8%和16.8%。
上述分析结果表明,Cas9对以上5个基因均实现了成功编辑,获得了不同突变类型的T0代植株,碱基突变类型主要以1个碱基的删除为主,约占总突变类型的50%以上,其次是2个碱基的删除以及多个碱基的删除,1个碱基的插入也是导致基因突变的重要因素。在各植株中仍然存在野生型基因序列,因此,T0代突变株为嵌合体。

图3 T0代植株中Cas9基因片段PCR电泳图Fig.3 PCR electrophoretogram of the sequence fragments of the Cas9 gene,DL 2000 Marker K326; Wild -type plant of K326;1~13,Transgenic plants

图4 靶位点序列的PCR电泳图Fig.4 PCR electrophoretogram of target sequence
对PCS1、eIF4E、TOM3、TOM1和HMA2基因在13棵T0代植株中的突变分布做了维恩分析,如图6所示,PCS1、eIF4E、TOM3、TOM1和HMA25个基因同时在7棵T0代植株中得到成功突变,这7棵T0代植株分别为 T0-4,T0-5,T0-6,T0-8,T0-9,T0-10和T0-11。5个基因在同一株烟草同时突变的检出率为53.8%。其余植株中均有不同基因突变的组合,且突变基因数目均在3个或3个以上,而单个基因突变的检出率在76.9%与92.3%之间,表明多基因敲除系统具有较高的编辑效率。

图5 Cas9介导烟草PCS1、eIF4E、TOM3、TOM1和HMA2基因突变注:红色为sgRNA序列,括号中分别标注了突变的碱基与对应的植株编号; WT:野生型Fig.5 Cas9-mediated targeted mutations of PCS1,eIF4E,TOM3,TOM1 and HMA2 in tobacco.Note:sgRNA sequence is marked in red,and the mutated bases and the corresponding plant number are marked in parentheses.WT:Wild Type

图6 PCS1、eIF4E、TOM3、TOM1和HMA2基因在T0代植株中的突变分布Fig.6 Mutation distribution of PCS1,eIF4E,TOM3,TOM1 and HMA2 in T0 Generation plants
2.6 脱靶位点预测与检测分析
由于与靶序列的相似,Cas9可能出现非特异性切割而发生脱靶现象。为了分析本文所构建的CRISPR/Cas9多基因编辑系统是否存在脱靶现象,我们将PCS1、eIF4E、TOM3、TOM1和HMA2基因的靶位点序列与烟草基因组数据进行了对比分析,根据((N)12NGG)的原则和PAM区上游序列(12 bp)决定靶位点特异性的原则[31,32],分别在烟草全基因组范围内各筛选了一个最有可能脱靶的候选位点,如表2所示,各基因靶位点从第8-10位点开始到PAM序列,完全与脱靶候选位点序列匹配。通过设计特异性引物,在5个基因都有突变的7株阳性植株中扩增候选脱靶位点的DNA片段,测序结果表明并未发现突变序列,这表明本实验所构建的CRISPR/Cas9系统具有较高的特异性。

表2 CRISPR/Cas9系统脱靶位点分析与检测Table 2 Analysis and examination of mutations in the putative off-target sites of CRISPR/Cas9 system.
3 讨论
CRISPR/Cas9基因编辑系统自出现以来,已在各植物中得到非常广泛应用。烟草是最早建立CRISPR/Cas9系统的作物之一,Gao等人最早在烟草中利用CRISPR/Cas9系统成功突变NtPDS和NtPDR6基因。随后,该系统在烟草腋芽、次生代谢、抗逆性等生物学性状的单基因功能研究方面发挥了重要作用[16-17,33-34]。本文基于烟草中已有CRISPR/Cas9基础表达系统,首次在烟草中针对不同生物学性状的多个基因开展了编辑研究。
构建了镉积累相关基因PCS1和HMA2、PVY抗性相关基因eIF4E、TMV抗性相关基因TOM3和TOM15个靶基因同时敲除的CRISPR/Cas9系统,并转化至烟草中。通过对阳性植株靶位点的基因序列分析表明,5个靶基因在烟草中均得到了成功编辑,5个基因同时突变的检出率为53.8%,该比例与水稻中对四个靶位点同时敲除的比例相当(44%和67%)[35],而单基因突变的检出率在76.9%与92.3%之间,这与烟草单基因突变的阳性植株检测率相当[16,17]。这表明本文构建的多靶点CRISPR/Cas9系统能够针对不同性状的不同基因进行编辑,为烟草不同优良性状的种质改良提供重要的技术依据。另外,由于普通烟草是由二倍体的绒毛状烟草和林烟草杂合而来的异源四倍体,在其基因组中有两套不同二倍体野生烟草基因组类型,因而存在同源基因数量多、基因功能冗余的现象。本文中构建的多靶点的CRISPR/Cas9系统,可有效应用于多个烟草基因的突变,提高烟草功能基因研究效率。
测序结果表明,CRISPR/Cas9系统在烟草中引起的突变,主要是1至10个碱基的删除,其中1个碱基删除的突变类型最多,这与前期在烟草和其他植物中所得出的结果一致,这种突变形式很大程度上与植物内在的修复机制相关。另外,由于普通烟草是由二倍体的绒毛状烟草和林烟草杂合而来的异源四倍体,基因组结构复杂且较大,在T0获得纯合突变的概率不高,多个基因的纯合突变筛选需更多的种植代数。CRISPR/Cas9系统引导核酸酶定向剪切活性存在一定程度的容错概率,会在目标位点序列相似的基因组区域存在非特异剪切活性(脱靶效应)。基因编辑靶位点预测方法较多,全基因组测序是目前被认为最有效的方法。对拟南芥、水稻、棉花中Cas9或Cpf1的脱靶效应研究发现,造成脱靶效应的最主要因素是gRNA特异性,sgRNA严谨性设计将有效消除脱靶效应的潜在影响[36-38]。本研究在5个基因同时发生突变的植株中,根据sgRNA特异性识别靶点原则预测和分析了可能的脱靶点,结果表明未发生脱靶现象,与前期在烟草中对CRISPR/Cas9脱靶检测结果一致[16]。这可能与目标基因的sgRNA均采用严谨标准有关,所设计靶序列均是全基因范围错误匹配最低以及GC含量合理的sgRNA。更全面的脱靶评估有待于后续进一步对纯合体突变体的全基因组测序分析研究。
4 结论
本研究针对烟草不同性状的基因构建了多靶点敲除的CRISPR/Cas9系统,获得了镉积累相关基因NtPCS1、HMA2、PVY抗性相关基因eIF4E以及TMV抗性相关基因TOM3、TOM1同时突变烟草遗传材料,5个基因同时突变的检出率为53.8%。脱靶效应评估分析显示,在所预测脱靶的候选位点上均未发生脱靶现象。本文构建的多基因编辑的CRISPR/Cas9系统可将烟草多个基因进行有效突变,为烟草基因功能的研究和基于CRISPR/Cas9技术的多性状改良奠定了基础。
猜你喜欢
奥秘(创新大赛)(2023年3期)2023-05-06 01:48:20
生物技术通报(2023年2期)2023-03-07 12:54:58
教学考试(高考生物)(2020年6期)2020-11-23 05:25:56
食品与生物技术学报(2020年8期)2020-01-06 08:00:56
科学24小时(2019年5期)2019-06-11 08:39:38
发明与创新(2019年9期)2019-03-26 02:22:48
浙江中西医结合杂志(2017年2期)2017-01-12 18:23:59
当代化工研究(2016年9期)2016-03-20 16:22:08
中国药业(2014年21期)2014-05-26 08:56:45
声屏世界(2014年6期)2014-02-28 15:18:09
