
赖氨酸特异性的去甲基化酶3A对内皮祖细胞功能的影响
2019-08-31 08:30柳小佩张博方陈静
中国心血管杂志 2019年4期
柳小佩 张博方 陈静
430060 武汉大学人民医院心血管内科
赖氨酸特异性的去甲基化酶3A[lysine(k)-specific demethylase 3A,KDM3A]是一种铁和草酸根依赖的双加氧酶,可特异性催化H3K9me1/2的去甲基化,具有表观遗传学特性[1]。前期研究证实,KDM3A在心血管疾病的发生发展中起重要的调控作用,可通过调控心肌炎症、凋亡、氧化应激关键因子的基因启动子区域的H3K9m2甲基化程度,调节相应关键因子表达,从而改变心肌细胞、血管平滑肌细胞等的多种功能[2-3]。
内皮祖细胞(endothelial progenitor cells,EPCs)在心血管疾病中的再生血管潜力已获广泛研究。EPCs是内皮细胞的主要来源,通过恢复内皮细胞的完整性和参与新生血管形成来修复血管损伤,同时也可通过旁分泌释放多种亲血管生成的分子来修复受损血管[4]。而KDM3A作为一种去甲基化酶,虽然调节多种细胞功能,但其调节EPCs的作用目前尚无充分研究结果。笔者推测,KDM3A可能参与调节EPCs的增殖、迁移及相关细胞因子的表达,从而发挥修复受损血管的功能。
1 材料与方法
1.1 试剂与仪器
活细胞计数试剂盒(cell counting kit-8,CCK-8)(日本Dojindo Laboratories公司),内皮细胞生长培养基(endothelial cell growth medium-2,EGM-2)(Lonza),胎牛血清(Gibco),一抗:甘油醛-3-磷酸脱氢酶(glyceraldehyde-3-phosphate dehydrogenase,GAPDH)(Abcam)、血管内皮生长因子(vascular endothelial growth factor,VEGF)(武汉三鹰)、基质细胞衍生因子1(stromal cell-derived factor-1,SDF-1)(Abcam)、磷脂酰肌醇3激酶/蛋白激酶B(protein kinase B,PI3K/Akt)和p-Akt(CST)。二抗:HRP-Goat Anti Rabbit(ASPEN)。酶标仪(美国Bio-Rad公司),荧光倒置显微镜(Nikon),Transwell小室(美国Corning公司)。
1.2 原代EPCs的分离与培养
用淋巴细胞分离液通过梯度离心法从同基因型成年雄性SD大鼠(体重150~180 g)的后肢长骨中分离出骨髓单核细胞(marrow mononuclear cells,MNCs)。随后,使用ECM-2分离并重悬于60 mm的细胞培养皿中,再加入20%血清、1%青霉素/链霉素,混匀,培养于37℃、95%空气、5%CO2的环境下。3 d后,待细胞形成集落,弃去培养基中的未贴壁细胞,再加入新配制的含有20%血清、1%青霉素/链霉素的细胞培养基,继续孵育4 d。7 d后,光镜下观察EPCs细胞呈纺锤形,聚集率达80%左右。将细胞用胰蛋白酶传代,培养3周后即出现典型的鹅卵石样原代EPCs。
1.3 原代EPCs的鉴定与实验设计
荧光标记的乙酰化低密度脂蛋白[1,1’-dioctadecyl-3,3,3’,3’-tetramethylindocarbocyanine (DiI)-labeled acetylated low density lipoprotein,Dil-acLDL]与FITC-UEA-1[the fluorescein isothiocyanate (FITC)-labeled Ulex europaeus agglutinin (UEA)-1]结合共染实验对EPCs进行鉴定[5]。将培养好的EPCs用PBS浸洗后加入Dil-acLDL(10 μg/ml),于37℃细胞培养箱中共培养4 h。4 h后再加入FITC-UEA-1(10 μg/ml),继续孵育1 h。孵育结束后,细胞进行4’,6-二脒基-2-苯基吲哚(4’,6-diamidino-2-phenylindole,DAPI)染色,在荧光显微镜下观察被Dil-acLDL和FITC-UEA-1双染的EPCs的数量。本实验采用3~5代的EPCs,按照完全随机设计的方法分为3组:(1)空白组(Con):EPCs培养3周后作为空白对照;(2)Ad-GFP转染对照组(Ad-GFP):使用空白腺病毒转染EPCs,作为病毒对照;(3)Ad-KDM3A转染处理组(Ad-KDM3A):使用含有KDM3A特异性腺病毒转染EPCs。
1.4 腺病毒的构建与转染
编码KDM3A的腺病毒载体(Ad-KDM3A)和对照所需的绿荧光蛋白(Ad-GFP)的腺病毒载体由上海吉凯公司设计和提供。本课题组既往研究显示MOI值为50时,腺病毒转染效率最佳[6]。当细胞生长达到培养皿的40%~50%时,根据最适MOI值转染相应的实验组,无血清培养基培养4 h后,弃去含有腺病毒的无血清培养基,更换正常培养基继续培养12 h,在荧光显微镜下观察细胞的转染效率。
1.5 细胞增殖的检测
用CCK-8试剂盒检测EPCs增殖能力。将各组培养皿中的细胞用胰蛋白酶消化,重悬接种入96孔板中,每组设置5个复孔。调整每孔的细胞数量为4×103个,每孔加入200 μl的EGM-2培养基后孵育24 h。孵育结束后每孔加入20 μl CCK-8试剂并继续培养4 h。最后使用酶标仪检测450 nm波长时各组的A值。
1.6 细胞迁移的检测
使用24孔、8 μm孔径的Transwell小室检测EPCs迁移能力。将各组细胞用胰蛋白酶消化后,调整每组细胞数量为105,用5%血清的EGM-2培养基重悬细胞后加入上室,在下室中加入20%血清的EGM-2。放入培养箱孵育12 h后,迁移的细胞用4%多聚甲醛固定30 min,再用0.1%结晶紫染色。在200倍显微镜下至少随机选取5个视野观察计数迁移的细胞数目。
1.7 细胞体外血管形成的检测
在96孔板的每孔中加入80 μl的基质胶,待1 h后基质胶凝固,将每组的EPCs数量调整为1×104,重悬于100 μl EGM-2培养基中,分别接种至已凝固的基质胶孔中。放入培养箱培养8 h,在光学显微镜下观察小管的结构并计算新生管腔数量。
1.8 细胞分泌能力及相关机制检测
用免疫印迹技术检测3组新生血管相关细胞因子VEGF、SDF-1和调控机制相关的Akt、p-Akt蛋白的表达。
1.9 统计学方法

2 结果
2.1 腺病毒转染效率干扰siRNA对KDM3A蛋白的下调作用
荧光显微镜下观察腺病毒的转染效率,腺病毒转染过的EPCs发出强烈的绿色荧光(图1A),本实验腺病毒在MOI=50时的转染效率接近90%。Western blot检测结果显示,Ad-GFP转染对照组的KDM3A蛋白表达水平明显高于Ad-KDM3A转染处理组(P<0.05),见图1B。

A:腺病毒转染过的EPCs发出强烈的绿色荧光(×40);B:腺病毒siRNA显著下调EPCs中KDM3A蛋白的表达,与空白组比较,aP<0.05图1 腺病毒的转染效率(n=3)
2.2 下调KDM3A的表达对EPCs的增殖能力的影响
相对于空白组,Ad-GFP转染对照组的EPCs在450 nm的A值比为1.031±0.059,空白组和Ad-GFP转染对照组组间比较无明显差异。而Ad-KDM3A转染处理组的EPCs的A值比为1.393±0.058,较Ad-GFP转染对照组明显增高(P=0.0016)。表明下调KDM3A蛋白的表达可以促进EPCs的增殖能力。
2.3 下调KDM3A的表达对EPCs的迁移能力的影响
相对于空白组,Ad-GFP转染对照组的EPCs迁移数量比值为1.21±0.11,空白组和Ad-GFP转染对照组组间比较无明显差异。而Ad-KDM3A转染处理组迁移至下室的EPCs数量比值为1.89±0.12,较Ad-GFP转染对照组明显增多(P=0.0023)(图2)。表明下调KDM3A蛋白的表达可以促进EPCs的迁移能力。
2.4 下调KDM3A的表达对EPCs体外血管形成能力的影响
相对于空白组,Ad-GFP转染对照组的新生血管管腔数量比为1.027±0.038,空白组和Ad-GFP转染对照组组间比较无明显差异。而Ad-KDM3A转染处理组为1.552±0.109,较Ad-GFP转染对照组明显增多(P=0.0103)(图3)。表明Ad-KDM3A转染处理组的体外血管形成能力更强,基本形成了完整的管腔。因此,下调KDM3A蛋白的表达可增强EPCs体外血管形成能力。
2.5 下调KDM3A的表达对EPCs分泌促血管生成因子能力的影响
Western blot检测结果显示,相比空白组,Ad-KDM3A转染处理组明显促进EPCs分泌VEGF和SDF-1(均为P<0.05),表明下调KDM3A的表达可以提高EPCs分泌促血管生成因子的能力。同时,还伴随着p-Akt/Akt蛋白表达的升高,提示KDM3A对VEGF、SDF-1分泌的调节可能是通过Akt信号通路发挥作用的(图4)。
3 讨论
冠心病作为最常见的心血管疾病,即使急性心肌梗死的患者在第一时间接受了冠状动脉旁路移植术和经皮冠状动脉介入治疗,还是会有部分患者术后复发,心肌再次缺血坏死甚至心力衰竭[7]。而改善心肌缺血的其中一种方法就是促进新生血管的形成来代偿心肌缺血造成的组织损伤[4]。在此过程中,心肌缺血损伤后,EPCs被认为是血管的“再生原料”迁移至缺血部位,通过形成毛细血管的结构成分和分泌血管生成因子来促进血管再生。而且EPCs也能分化为心肌细胞和平滑肌细胞,促进血管和心肌的修复,从而维持心肌梗死后的心功能[8-9]。因此,调控EPCs的生长和活性可能会缓解心肌缺血导致的一系列心血管疾病,如心绞痛、心肌梗死等,也可能改善心肌梗死患者预后。
KDM3A作为一种去甲基化酶,早期被证实可加重不同肿瘤细胞的增殖、迁移、侵袭及血管形成,其失调表达与多种肿瘤疾病相关,但后期研究表明KDM3A是一种多功能蛋白,具有高度调节亚细胞分布和基因转录作用。它可将多种环境因素与生理状态相结合,维持生物体内稳态[10-11]。而本课题组前期研究已证实了KDM3A在心血管疾病中的多种调控作用,通过下调KDM3A的表达,可显著改善心肌缺血再灌注损伤,故笔者推测KDM3A可能通过调控EPCs来发挥其相应的细胞功能。本研究结果表明,下调KDM3A的表达可显著增强EPCs的增殖、迁移、管形成功能,同时EPCs合成和分泌相关的促血管形成的细胞因子VEGF和SDF-1等分泌也有增加。因此,KDM3A具有调节EPCs的作用,可能为今后控制和防治心血管疾病提供新的思路。

图2 下调KDM3A蛋白的表达可以促进EPCs的迁移能力(n=3,×200)
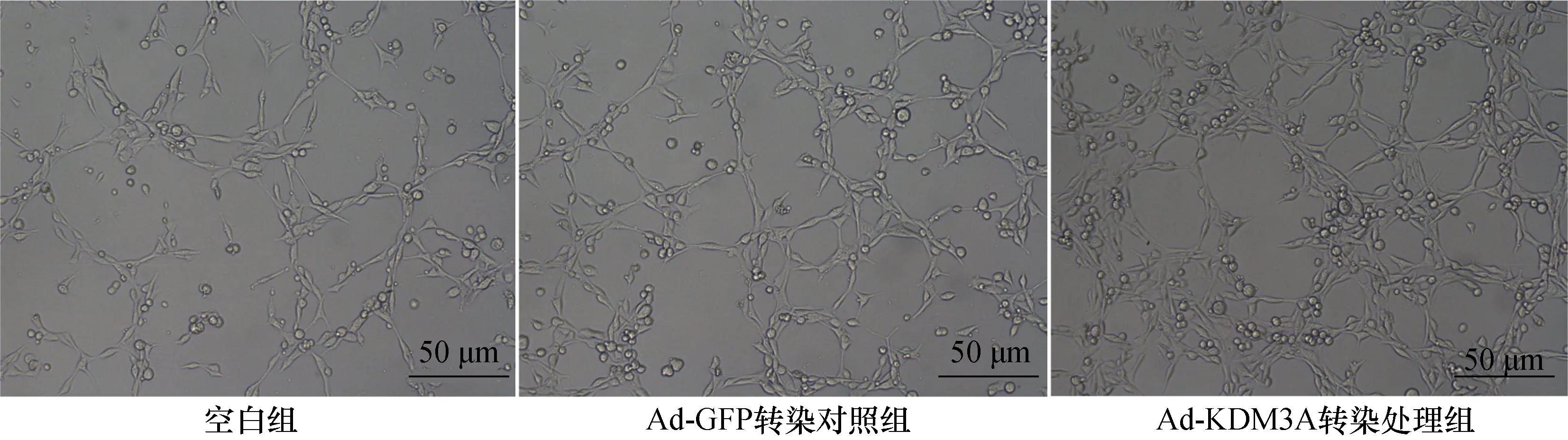
图3 下调KDM3A蛋白的表达可以增强EPCs体外血管形成能力(n=3,×200)
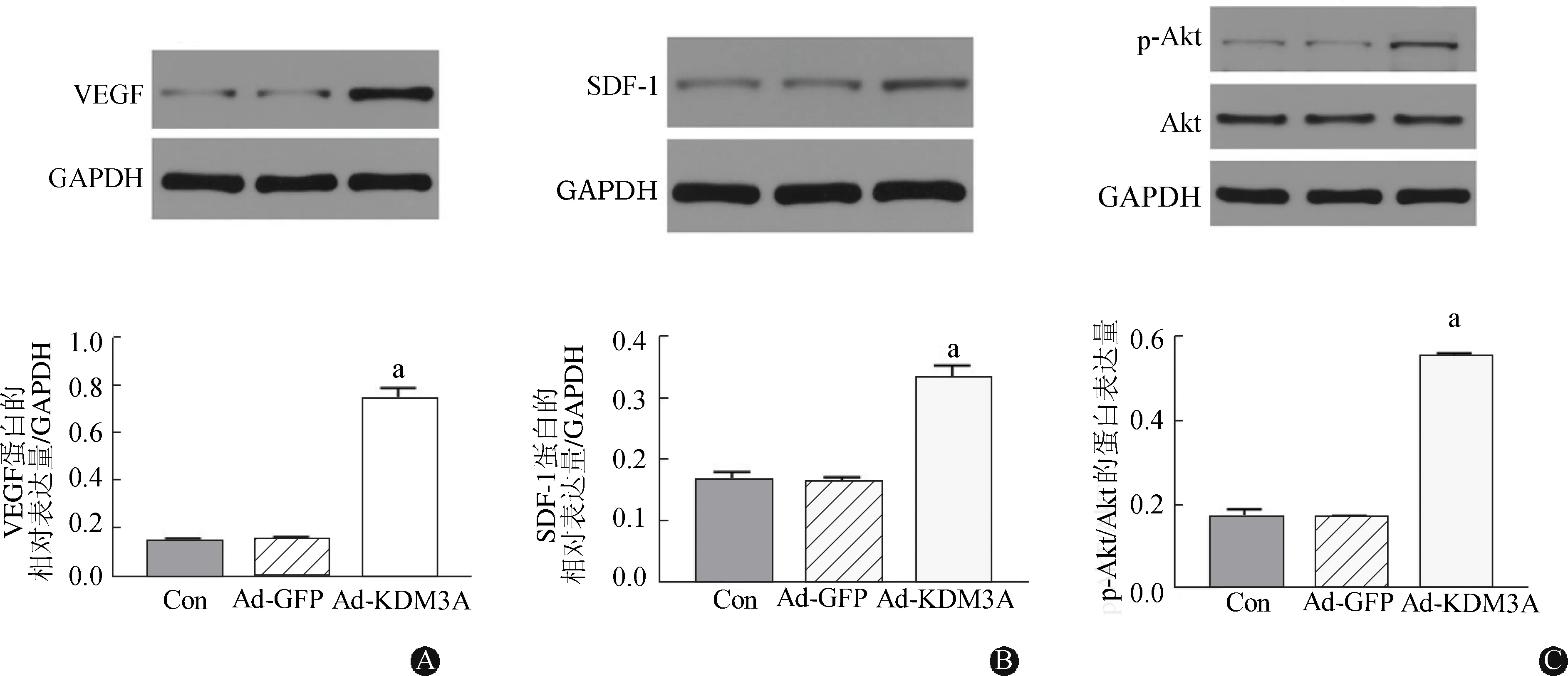
A、B:下调KDM3A蛋白的表达可以提高EPCs分泌促血管生成蛋因子VEGF、SDF-1的能力;C:下调KDM3A蛋白可以提高p-Akt蛋白的表达。与空白组比较,aP<0.05图4 下调KDM3A的表达对EPCs分泌促血管生成因子能力的影响(n=3)
Akt信号通路早期在肿瘤疾病中已有广泛研究[12]。癌细胞通过调节关键致癌信号级联,如通过PI3K/Akt信号级联通路来调控细胞内的氧化还原反应,从而调节癌细胞的增殖和活化[13]。而后期研究者们发现,Akt信号通路在癌症相关疾病和心血管疾病中的调控效应却截然相反。癌细胞持续的活化和增殖是治疗癌症所需要遏制的方向,但细胞生长、增殖和存活是治疗大多数心血管疾病的干预方向。Akt是心肌中最具特征的激酶之一,它通过旁分泌和自分泌因子的产生影响细胞功能的各个方面,包括生长、存活和增殖,以及代谢、葡萄糖摄取、基因表达和细胞间相互作用等,特别是作为各种血管生成细胞因子和生长因子的下游效应物[14-15]。Chu等[16]研究表明,抑制Akt磷酸化可改变骨髓干细胞的活性并抑制其内皮分化。本研究结果显示,下调KDM3A,EPCs各项检测指标增强的同时伴随着p-Akt/Akt蛋白表达的增高,进一步表明KDM3A调节EPCs活性与功能的机制可能与Akt蛋白有关。
综上,EPCs对于调节心肌缺血损伤、损伤修复、逆转心脏重构、维持心功能、内膜完整性、内皮功能和预防心血管并发症至关重要,而下调KDM3A蛋白的表达可增加EPCs增殖、迁移和管形成功能,同时促进EPCs分泌促血管形成相关的细胞因子,其机制可能与增加p-Akt/Akt蛋白表达有关。然而,本次研究仅仅在细胞水平上探讨了KDM3A对EPCs功能的调节,还需进一步经在体实验验证,同时除了Akt信号通路,潜在的其他相关下游信号通路也需进一步的研究和探索。
利益冲突:无
猜你喜欢
心血管病防治知识(2022年22期)2022-11-11
心血管病防治知识(2022年24期)2022-11-11
心血管病防治知识(2022年23期)2022-11-10
传染病信息(2022年3期)2022-07-15
现代临床医学(2022年3期)2022-06-06
中国动物保健(2022年2期)2022-05-05
养殖与饲料(2021年11期)2021-11-15
食品安全导刊(2020年21期)2020-12-03
食品安全导刊(2020年18期)2020-12-03
临床医药文献杂志(电子版)(2020年39期)2020-07-23
