
构建表达Flag-GPI的重组乙型肝炎病毒载体
2019-08-30 09:48胡孔颖谷陈建吴敏乐陶帅刘楠楠刘晶谢幼华
微生物与感染 2019年4期
胡孔颖,谷陈建,吴敏乐,陶帅,刘楠楠,刘晶,谢幼华
1. 复旦大学上海医学院基础医学院病原生物学系,教育部、卫健委、医科院医学分子病毒学重点实验室,上海 200032; 2. 上海市浦东医院,复旦大学附属浦东医院检验科,上海 201399
乙型肝炎病毒(hepatitis B virus,HBV)是重要的人类病原体,能感染人的肝实质细胞,引起急性和慢性肝脏炎症、肝纤维化、肝硬化和肝癌[1-6]。只有人类和少数灵长类动物的肝细胞对HBV具有易感性[7-8]。人肝细胞对HBV的易感性可归于多种因素,包括肝细胞表面的受体以及肝细胞内的蛋白和其他因子。早期的研究显示,肝细胞内富集的转录因子可调控HBV的基因表达。2012年肝细胞特异的牛磺胆酸钠共转运多肽(sodium taurocholate cotransporting polypeptide,NTCP)被鉴定为HBV的受体,极大地促进了对HBV易感性的研究[9-11]。在此基础上构建的过表达人NTCP的人肝癌细胞系HepG2-NTCP能够被HBV有效感染,为阐明HBV生活史提供了重要研究系统。但是在人肝癌细胞系Huh7细胞和未分化的HepaRG细胞中,过表达人NTCP后虽然可以造成HBV的感染,但是感染效率很低;在小鼠肝癌细胞系和大鼠肝癌细胞系如Hepa1-6、Hep56.1D和TC5123中,即使过表达人NTCP也不能使HBV有效感染[12-13]。并且,HBV感染HepG2-NTCP细胞系不仅要求有较高的感染复数(multiplicity of infection,MOI),还需要聚乙二醇(polyethylene glycol,PEG)和二甲亚砜(dimethyl sulfoxide,DMSO)的辅助[14]。因此,人肝细胞对HBV的易感性还需更深入的研究。
本课题组在前期研究中基于HBV临床突变株6898构建了具有较高复制能力的HBV载体5c3c。5c3c在包膜大蛋白preS1区缺失384bp,用以插入外源的基因片段,插入的外源基因可以利用其上游的Sp1启动子启动表达。此缺失部位包括preS1的C端和preS2的N端,位于不同开放读码框架的Spacer区域(图1B)[15-16]。此外,还将HBV包膜大蛋白preS1的起始密码子ATG突变为ACG,以避免preS1残留序列对下游插入的外源基因表达产生影响[15, 17]。本课题组前期在5c3c载体中插入了3种基因,分别是干扰素基因、shRNA的表达系统及ds-red基因,这些重组HBV载体不仅转染Huh7细胞后能表达外源基因,且保留有较高的复制能力,在回补HBV包膜蛋白的情况下,能够产生具有感染力的重组HBV(recombinant HBV,rHBV)[15]。
糖基磷脂酰肌醇(glycosylphosphatidylinositol,GPI)是一种蛋白质,通过翻译后修饰而在其C端获得的一个脂质锚,被修饰的蛋白质可以通过GPI锚定在细胞膜上,暴露于细胞表面[18-21]。能够获得GPI修饰的蛋白N端和C端各含有一个信号肽,N端信号肽的作用是将蛋白质带到内质网(endoplasmic reticulum,ER)内腔进行加工;C端信号肽又被称为GPI添加信号肽,其作用是用来替换预先在ER内合成的GPI,使GPI通过特定的化学键连接在蛋白质的C端[22-24]。
基于HBV载体5c3c和GPI的特性,本研究设想在5c3c载体中插入外源基因,该基因编码带有N端分泌信号肽和C端GPI添加信号肽的Flag标签,通过构建表达Flag-GPI的重组HBV,使Flag标签表达在感染细胞的表面,从而可以利用Flag抗体对感染细胞进行筛选,进而为HBV易感性提供研究工具[25-31]。
在本研究中,设计了两个将Flag序列插入5c3c中的方案。第1个方案中,在5c3c的插入位点依次插入自带起始密码子(ATG)的N端信号肽序列,Flag编序列以及GPI添加信号肽序列;第2个方案中,将5c3c 载体中preS1的ACG突变回ATG,称为5c3cT载体,利用preS1的N端充当信号肽,在5c3cT中顺序插入Flag和GPI添加信号肽序列。通过实验验证,最后得到重组HBV载体5c3cT-Flag-GPI和5c3c-CD59-Flag-GPI。它们转染Huh7细胞和HepG2-NTCP细胞后,可以将其表达的Flag锚定在细胞膜上。这两种重组HBV载体都保持一定的病毒复制能力,复制子5c3cT-Flag-GPI在回补HBV包膜蛋白的情况下,可以包装形成完整的重组HBV颗粒。
1 材料与方法
1.1 材料
1.1.1 细胞系293FT细胞系、Huh7细胞系由本实验室保存,HepG2-NTCP细胞系[带有灭瘟素(blasticidin S)抗性基因和人NTCP基因,在含blasticidin S的培养条件下能稳定表达人NTCP的HepG2细胞系]由王勇翔副教授惠赠。
1.1.2 质粒5c3c由本课题组保存,pCDNA3.1质粒由吴可菲硕士惠赠,pRRL-cPPT-PGK-GPI-scFv-AB65-HIS质粒由周保罗课题组惠赠,巨细胞病毒(cytomegalovirus,CMV)-1.1-HBV(由CMV启动子启动的1.1拷贝的HBV基因组)由本课题组保存,p-LMS(可以表达HBV大、中、小3种包膜蛋白的质粒)由崔晓娴博士惠赠。
1.1.3 仪器和试剂主要仪器包括PCR仪(北京东胜创新生物科技有限公司)、pH计(上海天达仪器有限公司)、二氧化碳细胞培养箱(Thermo公司)、EVOS倒置荧光显微镜(北京东胜创新生物科技有限公司)、Leica SP8 X激光共聚焦显微镜系统(德国Leica公司)、LSR Fortessa流式细胞仪(美国BD公司)。主要试剂包括PrimeSTAR Max DNA Polymerase(TaKaRa公司)、Go Taq qPCR Master Mix(Promega公司)、快速质粒小提试剂盒[天根生化科技(北京)有限公司]、普通琼脂糖凝胶DNA回收试剂盒[天根生化科技(北京)有限公司]、转染试剂TurboFect(Thermo Fisher Scientific公司)、PCR DIG Probe Synthesis Kit(Roche公司)、DIG Easy Hyb Granules(Roche公司)、DMEM(Gibco公司)、胎牛血清(Gibco公司)、Trypsin-EDTA(Gibco公司)、流式细胞抗体APC anti-Flag(Biolegend公司)、免疫荧光抗体anti-Flag(Sigma公司)、抗-preS1抗体(Santa Cruz Biotechnology公司)。
1.1.4 引物合成引物由上海桑尼生物科技有限公司合成,引物序列见表1。

表1 引物合成
1.2 方法
1.2.1 细胞培养细胞均在37 ℃ CO2培养箱中用DMEM(含10% FBS,100 U/ml青霉素和100 μg/ml链霉素)培养,HepG2-NTCP细胞的培养需要在培养基中另加blasticidin S(5 μg/ml)。
1.2.2 免疫荧光细胞用4%多聚甲醛室温固定30 min,PBS洗2次,每次5 min。加入含有1% Triton X-100的PBS破膜,室温孵育10 min。用含5% BSA、0.1% Triton X-100的PBS孵育细胞,进行封闭。弃去封闭液,用新配制的合适浓度的一抗孵育液(anti-Flag抗体按照1∶500~1 000 稀释)室温孵育2 h或4 ℃过夜。弃去一抗,PBS洗5次,每次5 min。配制合适浓度的二抗孵育液(一般按照 1∶500~1 000 稀释),室温孵育1 h。弃去二抗,PBS洗5次,每次5 min。加入DAPI稀释液,室温孵育2 min。弃去DAPI,PBS洗5次,每次5 min。最后封片,置于荧光显微镜下观察。检测细胞膜表面抗原时PBS中不加入Triton X-100,其他步骤相同。
1.2.3 流式细胞术(针对6孔板的贴壁细胞)弃去旧的培养基后用PBS轻轻清洗细胞2遍,加入400 μl胰酶稀释液(100 μl胰蛋白酶和300 μl PBS),37 ℃下消化5~10 min。加入800 μl DMEM(含10% FBS,100 U/ml青霉素和100 μg/ml链霉素)终止消化作用,重悬吹散后350 g离心5 min。用PBS重悬,对细胞进行计数后分装到1.5 ml的EP管中,使每管约含(5~10)×105个细胞。加入5 μl封闭液,室温封闭10 min。加入2 μl 0.2 μg/μl的APC anti-Flag抗体,冰上避光孵育20 min。用PBS洗2次,每次350 g离心5 min,收集细胞。用500 μl PBS重悬后上流式细胞仪分析。
1.2.4 Southern印迹法配制质量体积比为1%的琼脂糖凝胶,上样,70 V电泳约2 h。取出胶,用变性液变性1 h。完成后,用中和液中和1 h。结束后,将凝胶、裁剪好的尼龙膜、滤纸浸泡入20×SSC中。转膜过夜后,将尼龙膜在2×SSC溶液中浸泡5 min,接着紫外交联2 min。将尼龙膜放入圆柱形管中,加入5 ml的预杂交液, 42 ℃预杂交30 min。取出探针,100 ℃变性5 min,迅速冰浴3 min。将探针加入新的预杂交液中,配制形成杂交液。倒出预杂交液,换成杂交液, 42 ℃杂交6~8 h。高盐溶液洗膜2次,每次5 min。低盐溶液洗膜2次,每次15 min。接下来在杂交炉中的步骤温度都为 25~50 ℃。Washing缓冲液润洗1~5 min。Blocking 缓冲液封闭30 min。anti-DIG antibody溶液孵育30 min。Washing缓冲液洗2次,每次15 min。Detection缓冲液润洗2~5 min。在尼龙膜上覆盖CSPD工作液,室温避光5 min。发光,30 min以上。
2 结果
2.1 Flag-GPI表达载体的构建
通过GPI锚将蛋白锚定在细胞膜上并暴露于细胞膜外侧,需要目的蛋白(多肽)的N端和C端各含有一个信号肽。根据文献,选取了2个N端信号肽序列,分别命名为Bip和CD59。Bip信号肽序列来源于分泌表达载体pSecTag2A-Bip,其上Bip的作用是将所表达的蛋白引导入ER的分泌途径。CD59是补体调节蛋白CD59的N端信号肽,CD59本身通过GPI锚而被锚定在细胞膜表面[32]。C端的GPI添加信号肽的基因序列采用了周保罗课题组惠赠的载体质粒pRRL-cPPT-PGK-GPI-scFv-AB65-HIS上的GPI添加信号肽基因序列。每个N端信号肽都含有起始密码子(ATG),GPI添加信号肽的C端含有终止密码子(TGA)。
在真核表达载体pCDNA3.1中,选择在EcoRⅠ与BamHⅠ之间的酶切位点插入N端信号肽基因序列、Flag编码序列和C端GPI添加信号肽基因序列。为增强插入基因的翻译,在N端信号肽的起始密码子(ATG)前插入了Kozak序列(图1A)。同时,也构建了不含N端信号肽序列的对照质粒,即在pCDNA3.1载体中仅插入Flag编码序列和C端GPI添加信号肽,在Flag前端带有Kozak序列和起始密码子ATG(图1A)。以上这些质粒分别被命名为pCDNA3.1-Bip-Flag-GPI,pCDNA3.1-CD59-Flag-GPI和pCDNA3.1-Flag-GPI。
在含有1.1拷贝HBV基因组的5c3c载体质粒上进行了同样的操作,构建了5c3c-Bip-Flag-GPI和5c3c-CD59-Flag-GPI(图1C)。
5c3c载体上preS1仍保留其在N端的11个氨基酸的编码序列。HBV包膜大蛋白preS1的N端在天然情况下可被豆蔻酰化,因此可能含有将其定向带到ER的N端信号肽。基于该推断,将5c3c载体中preS1的突变起始密码子ACG变回ATG,新的载体命名为5c3cT。随后将Flag编码序列和GPI添加信号肽序列依次插入5c3cT载体中(图1C)。该载体质粒被命名为5c3cT-Flag-GPI。
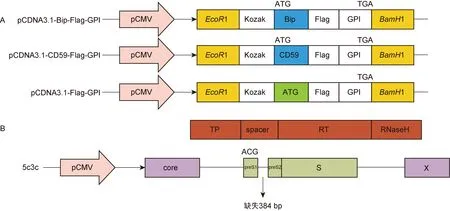
A: The sequences coding the N-signal peptide (Bip or CD59), Flag, and GPI-addition signal peptide (GPI) were inserted respectively into the vectors pCDNA3.1. B: The organization of the 5c3c vector. C: The sequences coding the N-signal peptide (Bip or CD59), Flag, and GPI-addition signal peptide (GPI) were inserted respectively into the vectors 5c3c and 5c3cT. The mutated preS1 start codon (ACG) in 5c3c was reverted to ATG in 5c3cT.
图1 Flag-GPI表达载体构建示意图
Fig.1 Schematic presentation of Flag-GPI constructs
2.2 Flag-GPI的细胞表面锚定验证
免疫荧光实验结果显示,pCDNA3.1-Bip-Flag-GPI和pCDNA3.1-CD59-Flag-GPI转染Huh7细胞(图2A)和293FT细胞(图2B)后,可将Flag标签锚定在细胞表面。阴性对照pCDNA3.1-Flag-GPI在细胞内可表达Flag,但不能将Flag标签锚定在细胞表面(图2A和2B)。通过流式细胞术进一步证实pCDNA3.1-Bip-Flag-GPI和pCDNA3.1-CD59-Flag-GPI转染Huh7细胞后,细胞表面Flag阳性的细胞数量显著增加(图2C)。
将5c3c-Bip-Flag-GPI、5c3c-CD59-Flag-GPI和5c3cT-Flag-GPI转染Huh7细胞后进行免疫荧光实验,发现5c3c-CD59-Flag-GPI转染的细胞表面可检测到Flag标签(图3A),而5c3c-Bip-Flag-GPI转染Huh7细胞后不表达Flag(结果未展示),5c3cT-Flag-GPI转染的Huh7细胞表面也呈现Flag阳性(图3A)。免疫荧光实验结果也证明,转染5c3c-CD59-Flag-GPI和5c3cT-Flag-GPI的 HepG2-NTCP细胞表面也能检测到Flag标签(图3B)。流式细胞检测结果进一步表明,5c3c-CD59-Flag-GPI和5c3cT-Flag-GPI转染的Huh7细胞可以将Flag标签锚定在细胞膜上(图3C)。
2.3 表达Flag-GPI的重组HBV具有复制能力
用DNA印迹法(Southern印迹法)验证获得的两个重组HBV复制子转染Huh7细胞后能否有效复制。实验结果表明,5c3c-CD59-Flag-GPI和5c3cT-Flag-GPI均能复制(图4)。如图4所示,在复制能力上空载体5c3c的最强,5c3cT-Flag-GPI与CMV-1.1-HBV相当,5c3cT-Flag-GPI比5c3c-CD59-Flag-GPI强。
由于5c3cT-Flag-GPI的复制能力比5c3c-CD59-Flag-GPI强,因此采用5c3cT-Flag-GPI进行进一步的实验。在Huh7细胞中共转表达HBV表面蛋白的p-LMS质粒和5c3cT-Flag-GPI质粒,纯化和浓缩细胞培养上清液后,用抗preS1的抗体进行pull-down实验,判断有无完整的重组HBV产生(图5)。共转p-LMS质粒和5c3c载体作为阳性对照,单转5c3c载体作为阴性对照。实验结果显示,5c3cT-Flag-GPI在回补HBV包膜蛋白后,可产生完整的重组HBV颗粒。
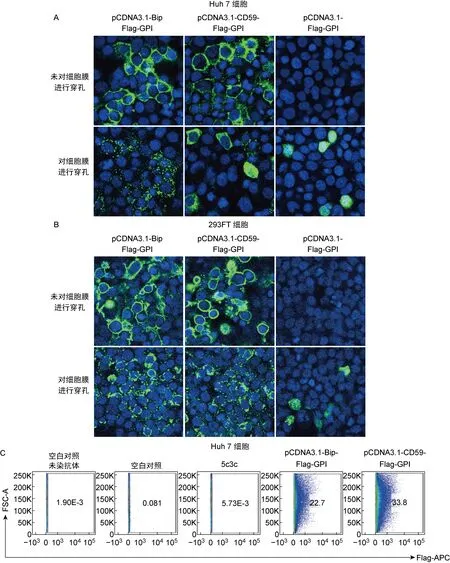
A: Transfections of pCDNA3.1-Bip-Flag-GPI and pCDNA3.1-CD59-Flag-GPI into Huh7 cells. pCDNA3.1-Flag-GPI served as negative control. B: Transfections of pCDNA3.1-Bip-Flag-GPI and pCDNA3.1-CD59-Flag-GPI into 293FT cells. pCDNA3.1-Flag-GPI served as negative control. C: Flow cytometry analysis. Surface-displayed Flag positive cells were detected and selected by anti-Flag antibodies.
图2 pCDNA3.1-Bip-Flag-GPI和pCDNA3.1-CD59-Flag-GPI转染细胞后可以将Flag标签锚定在细胞表面
Fig.2 Anchorage of Flag tags on the cell membrane after transfection of pCDNA3.1-Bip-Flag-GPI and pCDNA3.1-CD59-Flag-GPI
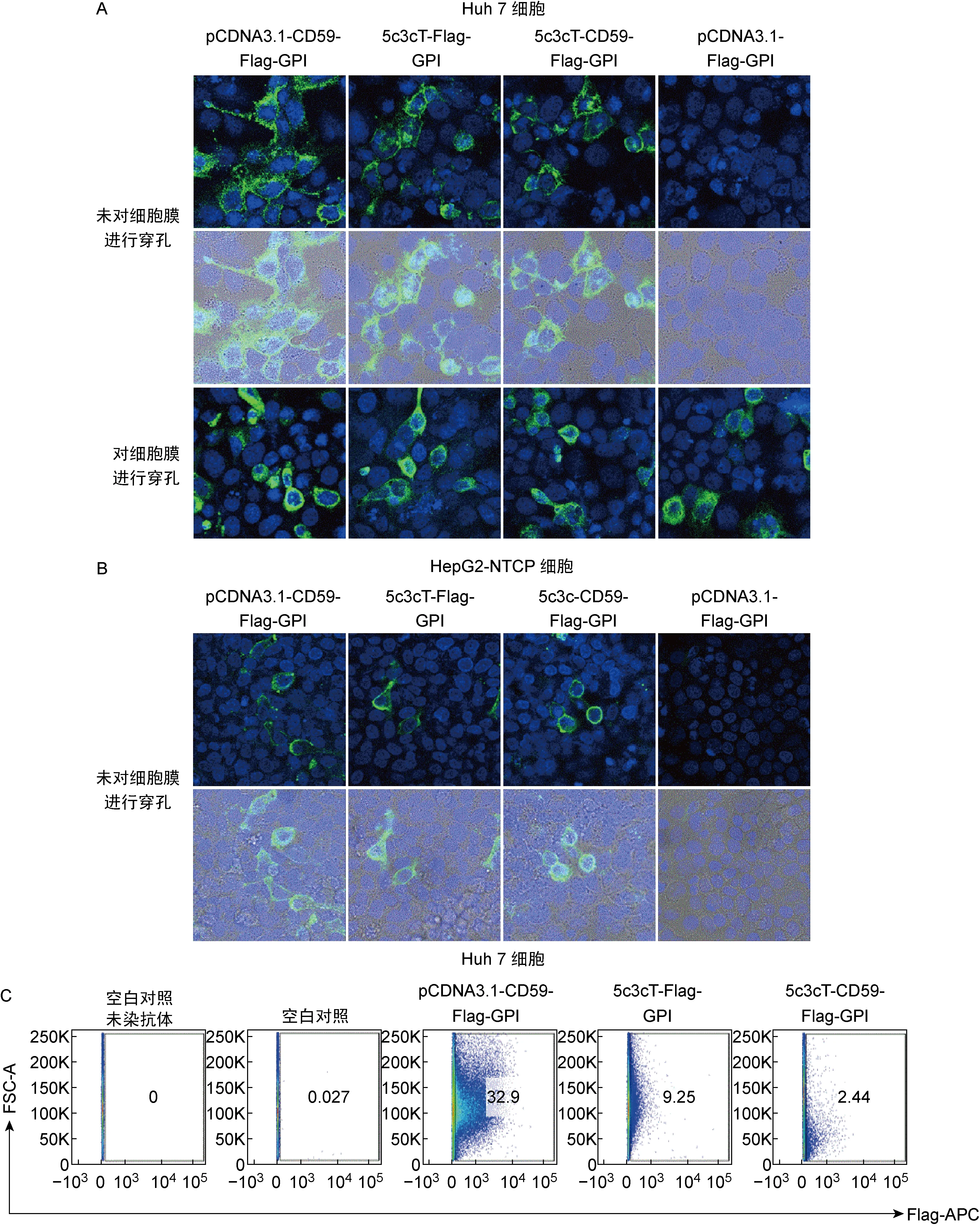
A: Transfections of 5c3cT-Flag-GPI and 5c3c-CD59-Flag-GPI into Huh7 cells. pCDNA3.1-Flag-GPI served as negative control. B: Transfections of 5c3cT-Flag-GPI and 5c3c-CD59-Flag-GPI into HepG2-NTCP cells. pCDNA3.1-Flag-GPI served as negative control. C: Flow cytometry analysis. Surface-displayed Flag positive cells were detected and selected by anti-Flag antibodies.
图3 5c3cT-Flag-GPI和5c3c-CD59-Flag-GPI转染细胞后可以将Flag标签锚定在细胞表面
Fig.3 Anchorage of Flag tags on the cell membrane after transfection of 5c3cT-Flag-GPI and 5c3c-CD59-Flag-GPI
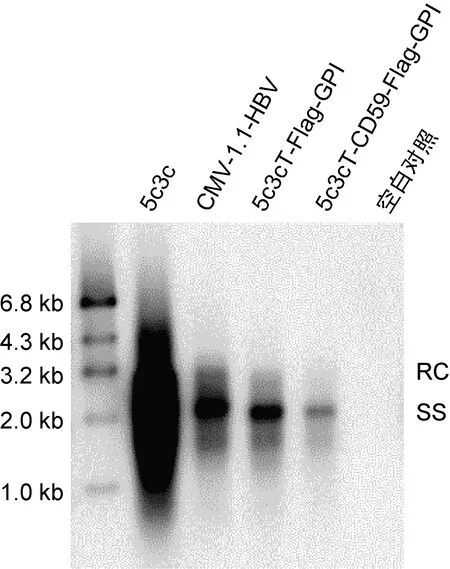
From the left to the right, the samples are 5c3c, CMV-1.1-HBV, 5c3cT-Flag-GPI, 5c3c-CD59-Flag-GPI, and blank control. 5c3c and CMV-1.1-HBV served as positive controls. Blank control served as a negative control.
图4 Southern印迹法验证5c3cT-Flag-GPI和5c3c-CD59-Flag-GPI的复制能力
Fig.4 The replication ability of 5c3cT-Flag-GPI and 5c3c-CD59-Flag-GPI verified by Southern blot
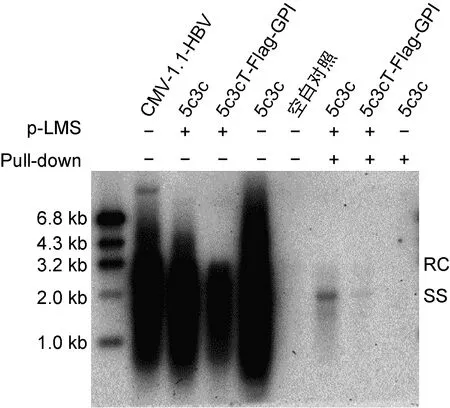
Cotransfection of 5c3c and p-LMS with pull-down assays served as positive control, transfection of 5c3c with pull-down assays served as negative control.
图5 Pull-down实验验证重组HBV的产生
Fig.5 Verification of formation of recombinant HBV particles with pull-down assays
3 讨论
在本研究中,首先在pCDNA3.1载体上获得pCDNA3.1-Bip-Flag-GPI和 pCDNA3.1-CD59-Flag-GPI,转染Huh7和293FT细胞后可将Flag标签锚定在细胞表面(图2)。这两个质粒转染293FT细胞后,进行对细胞膜穿孔和未穿孔的免疫荧光实验。结果显示,被穿孔的293FT细胞,细胞膜上有明显的穿孔痕迹,细胞膜表现为不连续的点状绿色荧光。推测与蛋白质相连的GPI锚(两个疏水的脂肪酸长链)大多插入在细胞膜上的脂质筏区域,该区域的稳定性较高,不溶于Triton X-100。因此在采用Triton X-100对细胞膜进行穿孔时,脂质筏区域不能被Triton X-100“穿透”,所以锚定有Flag而未被Triton X-100消化的脂质筏就呈现为不连续的点状绿色荧光[33-34]。然而对比观察pCDNA3.1-Bip-Flag-GPI和pCDNA3.1-CD59-Flag-GPI转染Huh7细胞后被Triton X-100穿孔后的免疫荧光结果发现,Huh7的细胞膜偶尔会出现不连续的点状绿色荧光,更多的是直接观察到细胞内核周围的绿色荧光,这与293FT细胞上所观察到的结果并不完全一致。原因可能是293FT细胞和Huh7细胞的细胞膜磷脂双分子中脂质筏的比例可能不一样, 293FT细胞膜磷脂双分子层中脂质筏的比例可能高于Huh7细胞膜中的,293FT细胞的细胞膜更难被Triton X-100消化,因此穿孔后293FT细胞的细胞膜更多呈现为不连续的点状绿色荧光。
pCDNA3.1-Bip-Flag-GPI 和 pCDNA3.1-CD59-Flag-GPI转染Huh7和293FT细胞后可以将Flag标签锚定在细胞膜上说明Bip和CD59可以正常发挥信号肽的功能,并且与Flag C端的GPI添加信号肽协同作用,协助将Flag标签锚定在细胞膜上。但是将这两个N端信号肽对应插入在载体5c3c上,就只有5c3c-CD59-Flag-GPI转染Huh7细胞后可以将Flag标签锚定在细胞膜上,而5c3c-Bip-Flag-GPI不能在转染的Huh7细胞中表达Flag标签(结果未展示)。此现象说明即使在pCDNA3.1载体上可以正常发挥作用的N端信号肽在5c3c载体上也不一定具有活性。可能是因为5c3c载体更为复杂,插入的N端信号肽在HBV基因组中发挥功能尚需具备一定的兼容性。本研究的目的之一是使Flag标签在转染细胞内可以通过GPI锚定在细胞膜上,但是否表达Flag标签的细胞都能有效地将Flag进行GPI修饰进而锚定在细胞膜上还需要进一步的研究,后续实验将检测Flag 膜锚定细胞数占Flag阳性细胞数的百分比,从而衡量GPI修饰效率和(或)Flag-GPI细胞膜锚定的效率。
载体5c3cT-Flag-GPI转染Huh7细胞后也可以将Flag标签锚定在细胞膜上,证明5c3c中残留的preS1序列确实具有N端信号肽的作用。HBV载体的相关研究也表明,HBV基因组的长度越短,该载体的复制能力则往往越强[16]。复制子5c3cT-Flag-GPI的基因组长度比复制子5c3c-CD59-Flag-GPI的短,但其复制能力比复制子5c3c-CD59-Flag-GPI的强,进一步验证了这一观点。
本研究基于课题组前期工作基础和GPI锚的相关特性,构建了重组HBV载体5c3cT-Flag-GPI和5c3c-CD59-Flag-GPI,转染Huh7细胞后可将Flag标签通过GPI锚定在细胞膜上,并可利用Flag抗体通过流式细胞术对表面含有Flag标签的细胞进行分选。5c3cT-Flag-GPI和5c3c-CD59-Flag-GPI转染Huh7细胞后都能进行复制,证明其都能产生完整的病毒核心颗粒。在反式回补HBV的包膜蛋白之后,5c3cT-Flag-GPI可以产生完整的重组HBV。因为产生的重组HBV滴度较低,所以重组HBV未能有效感染HepG2-NTCP细胞(结果未展示)。由图5可知在共转5c3cT-Flag-GPI和p-LMS未进行pull-down的情况下,复制子5c3cT-Flag-GPI的复制能力很强,说明此时可以产生大量的HBV核心颗粒;但是在共转5c3cT-Flag-GPI和p-LMS进行pull-down时,形成完整重组HBV颗粒的滴度却很低。该现象提示包装形成的重组HBV滴度不高的原因可能是重组HBV的包装系统不够高效。提示后续实验将改进包膜蛋白的提供方式来对重组HBV的包装体系进行优化,本课题组以往的包装系统采用反式提供HBV的包膜蛋白,但是包装效率不高。考虑到HBV的特性,顺式提供其包膜蛋白将是我们优化包装体系的一个重要研究方向。总的来说,本研究为进一步优化Flag-GPI重组HBV包装体系和感染实验奠定了基础。
猜你喜欢
福建农业学报(2022年3期)2022-05-24
廉政瞭望·下半月(2022年4期)2022-05-12
北华大学学报(自然科学版)(2021年3期)2021-07-13
今日农业(2021年2期)2021-03-19
心肺血管病杂志(2020年5期)2021-01-14
集装箱化(2019年7期)2019-10-18
江苏农业科学(2017年23期)2018-01-29
分析化学(2018年11期)2018-01-16
今日健康(2016年7期)2017-04-12
江苏农业科学(2016年5期)2016-07-23
