
AlN的MOCVD生长中表面吸附的量子化学研究
2019-08-22 07:50牛楠楠
人工晶体学报 2019年7期
牛楠楠,左 然
(江苏大学能源与动力工程学院,镇江 212013)
1 引 言
氮化铝(AlN)是重要的宽禁带半导体材料,在紫外LED和大功率电力电子器件中具有重要应用。AlN薄膜的主要生长技术是金属有机化学气相沉积(MOCVD),该技术不仅能够实现薄膜的大面积生长,还可以精确控制薄膜的组分和厚度。MOCVD生长AlN的关键是表面反应,即气相前体(如MMAl、NH3、DMAlNH2等)扩散到表面,发生吸附和脱附,并经过表面扩散和分解并入晶格。表面反应在很大程度上决定了薄膜的表面形貌、杂质组分和缺陷。AlN的生长目前仍存在生长速率低、表面形貌差等困难。为了制备出满足器件要求的高质量薄膜,深入了解AlN MOCVD表面反应机理至关重要。
前人已经针对III族氮化物MOCVD的表面反应做了大量研究。由于表面测量的困难,量子化学计算成为重要的工具。目前为止,相关研究大都是针对GaN生长的表面反应[1-7],针对AlN的表面反应研究目前还很少[8-13]。尽管AlN的表面反应与GaN相似,但由于Al-N键能(2.88 eV)比Ga-N键能(2.20 eV)高很多,造成含Al粒子在表面的吸附和扩散特性不同于含Ga粒子,因此AlN的表面反应机理与GaN相比仍有许多不同。Jindala等[8]利用密度泛函理论证明,AlN(0001)-Al面和AlN(000-1)-N面比其它表面更稳定。Akiyama等[9-10]利用第一性原理计算证明,Al原子在富N条件的吸附比富H容易得多,说明富N条件比富H条件更有利于AlN生长。Inagaki等[11]发现,AlN的生长速率主要受MMAl和DMAlNH2的影响,当DMAlNH2和AlN表面之间产生Al-N键时,吸附的DMAlNH2是稳定的。Suzuki等[12]证明,当以H2为载气时,N-H键的形成使AlN表面更稳定。Suzuki等[13]还研究了NH3在理想和H覆盖的AlN(0001)面的吸附,发现NH3不受表面H覆盖的影响,始终吸附在Top位;而NH2则分别吸附在Br位(理想表面)和Top位(0.75ML H覆盖表面)。
前人的研究主要考虑仅含Al-C键或N-H键的粒子(如MMAl、NH3等)在表面的反应,对于含有Al-N键的粒子(如DMAlNH2),Inagaki等[11]研究了DMAlNH2在AlN(0001)-Al面和-N面的吸附,但并没有给出所有的稳定吸附结构,且只考虑了理想表面。由于NH2是NH3分解吸附后的主要表面含N粒子[1,5-6],因此考虑吸附粒子在NH2覆盖面的吸附是必要的。本文利用量子化学的密度泛函理论(DFT),针对MOCVD-AlN的两种主要气相反应前体MMAl、DMAlNH2在理想和NH2覆盖的AlN(0001)-Al面的吸附进行优化计算。通过分析表面吸附位、吸附能、吸附前后分波态密度的变化等,确定可能的稳定吸附结构,从而进一步探讨AlN生长表面反应的机理。
2 计算模型
基于晶格参数为a=0.311 nm、c=0.498 nm的纤锌矿AlN[15],利用Material Studio软件,建立AlN(0001)-Al面的2×2周期超晶胞模型,如图1(NH2覆盖表面类似)。该模型纵向共7层原子,固定底部3层原子,表面原子充分弛豫,采用H原子钝化底部悬挂键[6]。真空层高度取2 nm,使材料表层电荷密度在真空区域逐渐消失为零,以保证上下表面之间的相互作用可忽略[16]。为了简化计算,对于NH2覆盖表面,假设NH2表面覆盖度为1ML,且NH2均吸附在Top位。
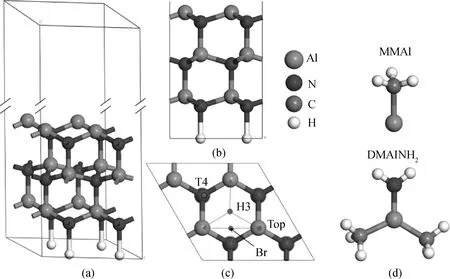
图1 AlN(0001)-Al面2×2周期超晶胞模型的三维视图(a)、主视图(b)和俯视图(c);(d) MMAl、DMAlNH2的分子模型Fig.1 3D(a), front(b), and top(c) views of the 2×2 periodic supercell model of AlN(0001)-Al surface; (d) Molecular models of MMAl and DMAlNH2
所有计算均在CASTEP程序包中进行。其中,电子交换-关联能的计算采用广义梯度近似GGA和PW91泛函相结合的方法[17-20],因为它们能够更准确地描述电子密度迅速变化的化合物半导体表面。为了使平面波基组计算中所采用的截止能尽可能小,离子实与价电子之间的相互作用采用超软赝势。考虑到计算精度和计算成本,描述体系电子波函数的平面波基组的截断动能取300 eV,k点设置为4×4×1,此设置精度足够满足AlN几何优化和能量优化需求。
理想AlN(0001)-Al面超晶胞模型如图1(a~c)所示,每个表面Al原子均具有一个悬挂键,各包含3/4个电子,它能够与吸附粒子中的未饱和键形成共价键。MMAl、DMAlNH2的分子结构如图1(d)。二者中的CH3基团的电子已饱和,不能与表面成键。三价的Al原子和五价的N原子均可以通过轨道杂化而与表面成键。两种分子吸附在表面后,最终都脱去甲基[11],形成sp3杂化键的AlN晶体。
本文针对气相粒子MMAl、DMAlNH2在理想和NH2覆盖的AlN(0001)-Al面的吸附进行DFT计算。首先优化吸附前的气相粒子以及AlN表面。然后将优化后的吸附粒子放置于表面上方,与表面一起优化。通过计算和对比吸附粒子的吸附位、吸附能、分波态密度图等,寻找AlN薄膜的表面生长机理。在此基础上,对NH2覆盖的AlN表面进行同样优化计算和分析。
吸附能△Eads定义为吸附前后系统总能量的变化[9,21]:
△Eads=Eads/sur-(Eads+Esur)
(1)
其中Eads/sys为吸附后系统的总能量,Eads和Esys分别为吸附前的吸附粒子和系统的总能量。若吸附能为负值,则表示发生吸附,能量越负吸附趋势越大。
3 结果与讨论
3.1 理想AlN(0001)-Al面吸附
利用前述的优化计算方法,在理想AlN(0001)-Al面上方将MMAl垂直放置(Al原子向下,向上则不能成键)、DMAlNH2水平放置,得出MMAl、DMAlNH2的稳定吸附结构,如图2所示。其中下标ads和s分别代表吸附原子和表面原子。
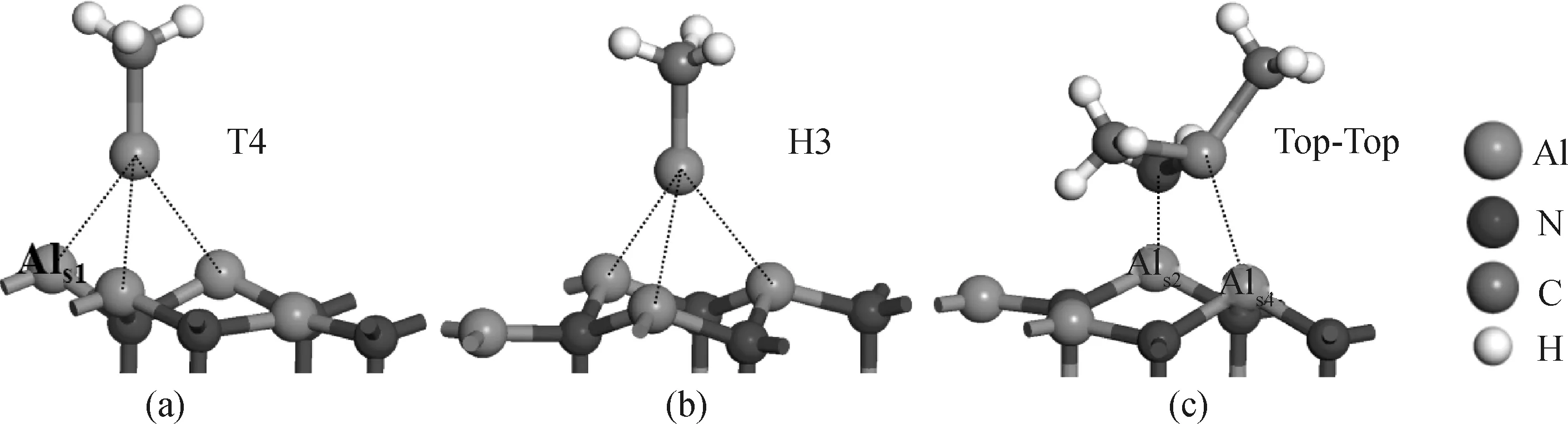
图2 MMAl、DMAlNH2在理想AlN(0001)-Al面的吸附结构:(a)和(b) MMAl分别吸附在T4位和H3位,与表面形成3个Alads-Als键;(c) DMAlNH2吸附在Top-Top位,与表面形成Nads-Als2键和Alads-Als4键Fig.2 Adsorption structure of MMAl and DMAlNH2 on the ideal AlN(0001)-Al surface: (a) and (b) MMAl adsorbed at the T4 and H3 sites, respectively, with three Alads-Als bonds; (c) DMAlNH2 adsorbed at the Top-Top site with Nads-Als2 and Alads-Als4 bonds
MMAl有两种吸附结构,即分别吸附在表面的T4位和H3位(如图2(a)、(b)),与表面形成3个Alads-Als键,键长分别为0.273 nm、0.273 nm、0.298 nm(T4位)和0.278 nm、0.279 nm、0.290 nm(H3位)。两种结构的吸附能分别为-2.89 eV、-2.94 eV,大小相近,说明MMAl在T4位和H3位吸附概率也相似。DMAlNH2吸附前与表面平行,吸附后一个CH3与表面原子互相排斥而远离表面,如图2(c)。DMAlNH2吸附在表面的Top-Top位,粒子中Al原子和N原子分别与表面最近邻的两个Al原子形成Alads-Als4键和Nads-Als2键(与DMGaNH2的吸附类似[2]),键长分别为0.262 nm和0.192 nm,与文献值(Al-Als键:0.267 nm[9];N-Als键:0.190~0.210 nm[13])相近。吸附能为-3.35 eV,低于MMAl,说明DMAlNH2吸附趋势更大。
为进一步分析MMAl、DMAlNH2在理想AlN表面的吸附,本文计算了吸附前后成键原子的分波态密度(PDOS)。图3示出MMAl在理想AlN表面T4位吸附前后的PDOS图。因参与成键的3个表面Als原子的PDOS相似,故图中只显示了一个表面Als1的状态。
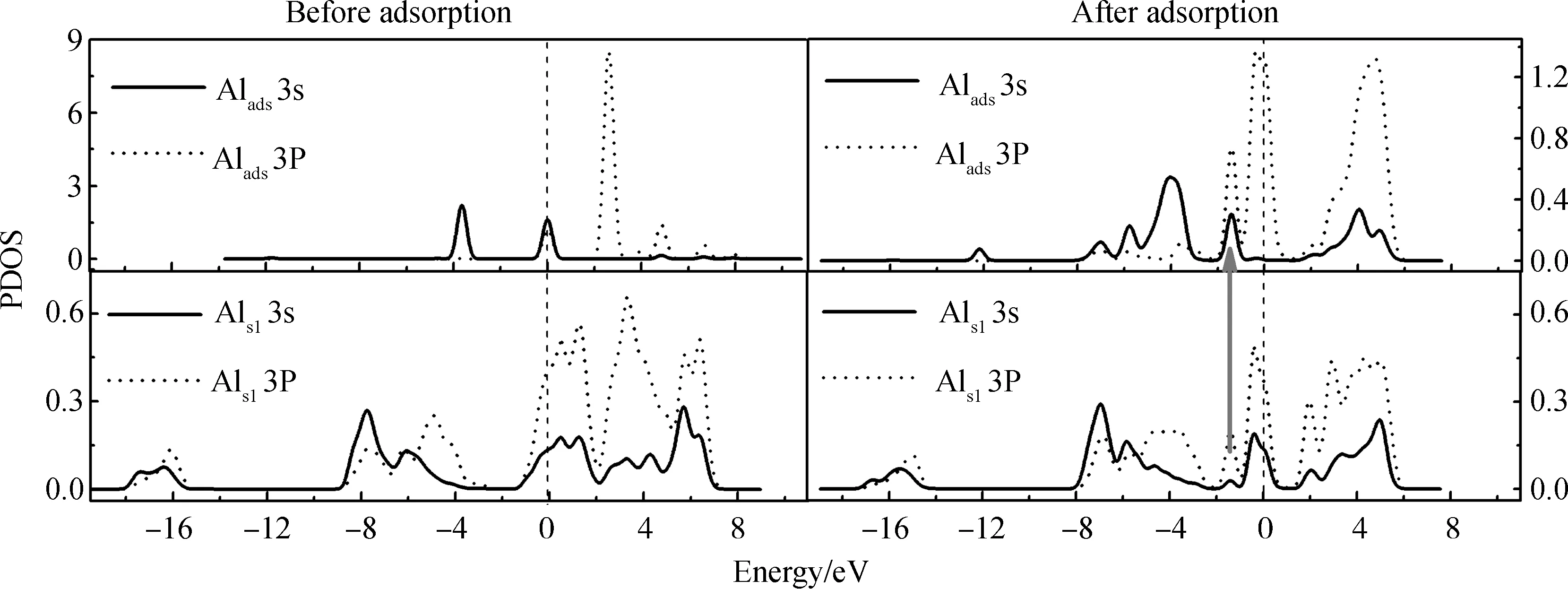
图3 在理想AlN表面,MMAl在T4位吸附前后成键原子的分波态密度(PDOS)图(图中箭头标注出部分共振)Fig.3 PDOS of the bonding atoms before and after MMAl adsorption at T4 site on ideal AlN surface (The arrow in the figure indicates part of the resonance)
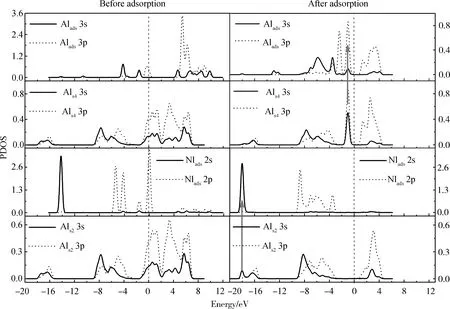
图4 在理想AlN表面,DMAlNH2吸附前后成键原子的分波态密度(PDOS)图(图中箭头标注出部分共振)Fig.4 PDOS of the bonding atoms before and after DMAlNH2 adsorption on ideal AlN surface (The arrow in the figure indicates part of the resonance)
从图3可看出,吸附前Alads原子的3s、3p轨道峰值尖锐,显示出较强的局域性,无明显的相互作用;其它Al原子的3s、3p轨道在多个能量区间内均存在大面积的重叠,说明这些原子发生了sp轨道杂化。对比吸附前后Alads原子的态密度可以看出,吸附后Alads原子的3s、3p轨道均向左移动,同时峰值明显降低且能带变宽,说明吸附后Alads原子的3s、3p轨道发生电荷转移并重新分布,系统能量降低。吸附后,Alads原子的3s、3p轨道与Als1原子的3s、3p轨道均有一定的共振,说明吸附原子通过sp杂化与表面原子相互作用,证明了Alads-Als键的存在。MMAl在H3位吸附的PDOS图(本文省略)与在T4位吸附相似,说明MMAl在H3位吸附同样形成Alads-Als键。
图4所示为DMAlNH2在理想AlN表面Top-Top位吸附前后成键原子的PDOS图。从图4中可看出,吸附前后DMAlNH2中Alads原子和表面Als原子的3s、3p轨道均在多个能量区内存在重叠,说明它们都发生了sp轨道杂化。对比吸附前后Alads和Nads原子的态密度可看出,吸附后二者的s、p轨道均向左移动,同时峰值明显降低且能带变宽,说明吸附后Alads和Nads原子的s、p轨道发生电荷转移并重新分布,系统能量降低。DMAlNH2吸附后,Alads原子的3s、3p轨道与Als4原子的3s、3p轨道均有明显共振,证明了Alads-Als4键的存在;同样,Nads原子的2s轨道与Als2原子的3s轨道也发生共振,证明了Nads-Als2键的存在。
3.2 NH2覆盖AlN(0001)-Al面吸附
与3.1类似,首先对NH2覆盖的AlN表面进行优化计算。然后将MMAl垂直放置(Al原子向下)、DMAlNH2水平放置(Al位于Top位)于NH2覆盖的AlN表面上方,进行优化计算。得到MMAl、DMAlNH2的稳定吸附结构,如图5所示。
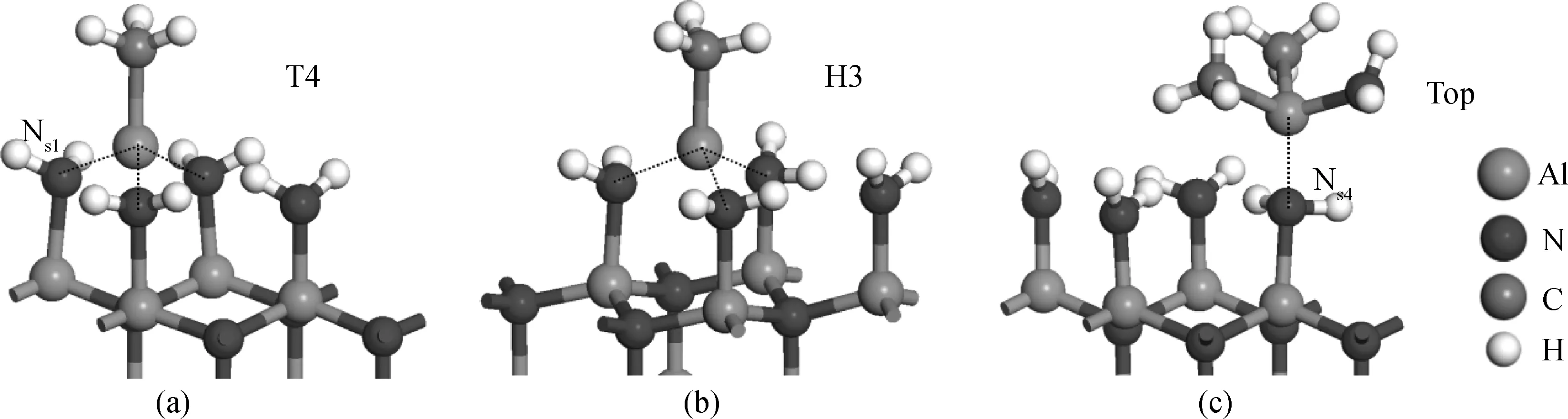
图5 MMAl、DMAlNH2在NH2覆盖AlN(0001)-Al面的吸附结构;(a)和(b) MMAl吸附在T4位和H3位,与表面形成3个Alads-Als键;(c) DMAlNH2吸附在Top位,与表面形成Alads-Ns4键Fig.5 Adsorption structure of MMAl and DMAlNH2 on NH2-covered AlN(0001)-Al surface: (a) and (b) MMAl adsorbed at T4 and H3 sites with three Alads-Als bonds formed; (c) DMAlNH2 adsorbed at Top site, with one Alads- Ns4 bond formed
与MMAl在理想表面的吸附相似,MMAl在NH2覆盖AlN(0001)-Al面也有两种吸附结构,即T4位和H3位,分别与表面形成3个Alads-Ns键(如图5(a)、(b)),键长分别为0.190 nm、0.190 nm、0.191 nm(T4位)和0.189 nm、0.190 nm、0.192 nm(H3位),与AlN晶格的Al-N键长相近(0.191 nm[9])。两种结构的吸附能(分别为-5.52 eV、-5.47 eV)也相近,说明MMAl在T4位和H3位的吸附概率相近。
DMAlNH2在NH2覆盖AlN(0001)-Al面只有一种稳定吸附结构,即其中的Alads原子吸附在Top位,与表面Ns原子形成一个Alads-Ns键(如图5(c)),而NH2和CH3遭到排斥向远离表面的方向翘起。Alads-Ns键长为0.198 nm,与文献值相近(0.190 nm[13])。吸附能为-0.34 eV,明显小于MMAl的吸附能,说明在NH2覆盖AlN(0001)-Al面,MMAl的吸附趋势比DMAlNH2大得多。
为进一步分析MMAl、DMAlNH2在NH2覆盖AlN表面的吸附,计算了吸附前后成键原子的PDOS。图6所示为MMAl在T4位吸附前后的PDOS图。与图3类似,图6也只显示了一个表面原子Ns1。从图中可看出,Alads原子吸附前的3s、3p轨道峰值尖锐,显示出较强的局域性,无明显的相互作用;吸附后的3s、3p轨道在多个能量区内均存在重叠,说明它们发生了sp轨道杂化。对比吸附前后Alads原子的态密度可看出,吸附后Alads原子的3s、3p轨道均向左移动,同时峰值明显降低且能带变宽,说明吸附后Alads原子的3s、3p轨道发生电荷转移并重新分布,系统能量降低。MMAl吸附后,Alads原子的3s、3p轨道与Ns1原子的2s、2p轨道均有明显共振,说明吸附原子通过sp杂化轨道与表面原子相互作用,证明了Alads-Ns键的存在。MMAl在H3位吸附的PDOS图(本文省略)与在T4位吸附相似,说明MMAl在H3位同样形成Alads-Ns键。
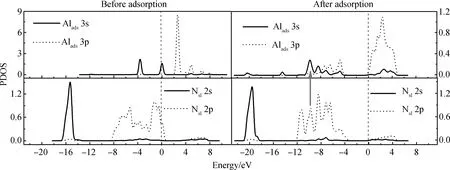
图6 在NH2覆盖的AlN表面,MMAl在T4位吸附前后成键原子的分波态密度(PDOS)图(图中箭头标注出部分共振)Fig.6 PDOS of the bonding atoms before and after MMAl adsorption at T4 site on NH2-covered AlN surface (The arrow in the figure indicates part of the resonance)
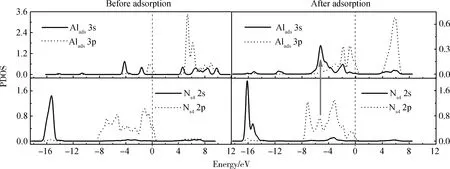
图7 在NH2覆盖的AlN表面,DMAlNH2吸附前后成键原子的分波态密度(PDOS)图(图中箭头标注出部分共振)Fig.7 PDOS of the bonding atoms before and after DMAlNH2 adsorption on NH2-covered AlN surface (The arrow in the figure indicates part of the resonance)
图7所示为NH2覆盖AlN表面,DMAlNH2在Top位吸附前后成键原子的PDOS图。从图中可看出,Alads原子的3s、3p轨道在多个能量区间内均存在重叠,说明它们发生了sp轨道杂化。对比吸附前后Alads原子的态密度可以看出,吸附后Alads原子的3s、3p轨道均向左移动,同时峰值明显降低且能带变宽,说明吸附后Alads原子的3s、3p轨道发生了电荷转移并重新分布,吸附后系统能量降低。DMAlNH2中Alads原子的3s、3p轨道与表面Ns4原子的2s、2p轨道均有一定的共振,说明它们形成了Alads-Ns4键。
4 结 论
利用量子化学的密度泛函理论,计算了AlN的MOCVD生长中气相前体MMAl、DMAlNH2在理想和NH2覆盖AlN(0001)-Al面吸附的吸附位、吸附能、分波态密度等。计算结果表明:在理想和NH2覆盖的AlN表面,反应前体MMAl吸附在T4位和H3位,MMAl中的Al原子与表面原子形成3个Alads-Als键(理想AlN表面)或3个Alads-Ns键(NH2覆盖的AlN表面)。在任一表面,MMAl在T4位和H3位吸附的吸附能均大小相近,说明吸附概率也相近。在理想AlN表面,DMAlNH2吸附在Top-Top位,与表面最近邻的两个Al原子分别形成Alads-Als4键和Nads-Als2键,吸附能大于MMAl,说明DMAlNH2优先吸附;在NH2覆盖的AlN表面,DMAlNH2吸附在Top位,与表面最近邻的N原子形成Alads-Ns键,吸附能明显小于MMAl,故MMAl优先吸附(注:这里吸附能考虑绝对值)。
猜你喜欢
北京航空航天大学学报(2022年7期)2022-08-06
都市人(2022年3期)2022-04-27
昆明医科大学学报(2022年1期)2022-02-28
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
国际太空(2021年8期)2021-11-05
新疆大学学报(自然科学版)(中英文)(2020年2期)2020-07-25
环球时报(2019-12-05)2019-12-05
