
Leptin-induced Notch and IL-1 signaling crosstalk in endometrial adenocarcinoma is associated with invasiveness and chemoresistance
2019-08-22 01:53DanielleDaleyBrownAdrianaHarbuzariuAnnAnuKurianGabrielaOpreaIliesRubenReneGonzalezPerez
Danielle Daley-Brown, Adriana Harbuzariu, Ann Anu Kurian, Gabriela Oprea-Ilies, Ruben Rene Gonzalez-Perez
Abstract
Key words: Endometrial cancer; Leptin; Notch; Interleukin-1; Notch, IL-1 and Leptin crosstalk outcome; Chemoresistance
INTRODUCTION
Endometrial cancer (EmCa) accounts for 7% of new cancer cases among women in the United States. In 2017, approximately 61400 EmCa cases have been reported with about 11000 women that succumbed to the disease[1]. Obesity is a major risk factor for EmCa[2,3]. Indeed, the incidence of EmCa is increasing in developing countries where obesity is on the rise[4]. Obese individuals exhibit elevated serum levels of the adipokine leptin. Leptin's primary role is to signal as a long term satiety factor that helps to maintain the food intake and energy balance[5]. However, leptin signaling controlling appetite is impaired in obese individuals, which suggests a kind of leptinresistance status[5]. Leptin is mainly secreted by adipocytes but also by the gastric mucosa, heart, placenta, and skeletal muscle. In addition, cancer cells secrete leptin that induces tumor proliferation, inflammation and angiogenesis in various cancer types[6,7].
Previous studies have demonstrated that leptin up-regulates various signaling pathways involved in cancer progression[5]. Studies have shown that leptin upregulates Interleukin-1 (IL-1) signaling in breast and EmCa cells, and Notch in breast and pancreatic cancer cells[8-12]. In particular, leptin-induced Notch and IL-1 pathways are associated with breast cancer cell proliferation, migration, invasion as well as chemoresistance[7,10]. Notch signaling is an evolutionarily conserved pathway that affects cell differentiation, proliferation and apoptosis across various cell types at different stages of development[5]. Consequently, mutations of Notch genes can lead to many diseases within organs and tissues[13]. Likewise, IL-1 is known to be upregulated in many tumor types, where promotes tumor angiogenesis, growth and metastasis[14]. A novel signaling crosstalk between leptin, Notch and IL-1 (NILCO) was associated with breast cancer progression[10].
Chemotherapy is a standard therapy in multiple cancers. While chemotherapy is often capable of inducing apoptosis in cancer cells that eventually leads to a reduction in the tumor size, cancer recurrence is still a major issue. Recurrence rates often result in death because of treatment failure[15].
NILCO expression was previously found in human EmCa[16], but whether leptin can regulate its expression and whether is involved in EmCa proliferation, aggressiveness and chemoresistance remain to be determined. We hypothesize that NILCO is essential in the regulation of leptin-mediated induction of proliferation, migration/invasion and chemoresistance in EmCa cells, and that leptin's effects will be more evident in the more aggressive and poorly differentiated EmCa cells. In the present study, we found that leptin mediated the expression of Notch and IL-1 signaling components, which was higher in type II EmCa cells. In addition, NILCO impacted on the proliferation and invasiveness of type I and II EmCa cells. Moreover, leptin impaired paclitaxel cytotoxic effects on EmCa cells.
MATERIALS AND METHODS
EmCa cell lines and reagents
EmCa cell lines HEC-1A, KLE and An3Ca, and 1% penicillin-streptomycin solution were purchased from American Type Culture Medium (ATCC). EmCa Ishikawa cell line, monoclonal antibodies anti-Notch1 and GAPDH, dimethyl sulfoxide (DMSO)and other common reagents were obtained from Sigma (St. Louis, MO). Polyclonal Notch4, JAG1, IL-1R tI, and OB-R antibodies were obtained from Santa Cruz Biotechnology. Polyclonal anti-Notch2, Notch3 and DLL4 antibodies were purchased from Abcam. Monoclonal survivin antibody was obtained from Cell signaling, and anti-Hey2 antibody was purchased from Millipore. Vybrant 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) proliferation kit was from LifeTechnologies (Grand Island, NY). Cell Counting Kit- 8 (CCK-8) was from Dojindo Molecular Technologies Inc. (Japan). Leptin was purchased from R and D Systems(Minneapolis, MN). The leptin peptide receptor antagonist 2 (LPrA2) was synthetized and purified as previously described[17]. LPrA2 was bound to iron oxide nanoparticles(IONP; Ocean Nanotech, San Diego, CA) as described elsewhere[18,19]. Specific small interfering RNA (siRNA) for Notch1, Notch3 and Notch4 were obtained from(Qiagen). Annexin V/ Fluorescein Isothiocyanate (FITC) and Propidium Iodide (PI)for cell cycle analysis were obtained from Nexcelom Bioscience Boston, MA. Paclitaxel(PTX) was obtained from SelleckChem Houston, TX.
Cell cultures
Type I (Ishikawa, and HEC-1A) and type II EmCa cells (An3Ca and KLE) were cultured in Dulbecco's Modified Eagle's medium (DMEM), 10% fetal bovine serum(FBS from Gemini Bioproducts) and 1% penicillin-streptomycin (ATCC) until they were 80% confluent. Then, cells were serum-deprived by culturing in basal medium for 24 h, and treated for additional 24 h in basal medium containing increasing leptin doses (0, 0.6, 1.2, and 6.25 nM, equivalent to 0, 10, 20 and 100 ng/mL, which characterized leptin levels in overweight, obese and morbid obese patients,respectively) and inhibitors of leptin (IONP-LPrA2), Notch (siRNA Notch1, Notch3 and Notch4) and IL-1 signaling (antibody anti-IL1 R tI) or with the chemotherapeutic paclitaxel.
Western blotting
Expression levels of Notch receptors, ligands and targets were determined by Western blotting (WB) analysis in EmCa cells cultured under different treatments. Cellular protein content after treatments was obtained using RIPA lysis buffer. Protein concentrations of cell lysates were determined using the Bradford Assay. Membrane blocking was performed using 5% non-fat milk in TBST for 30 min at room temperature. A 1:200 dilution of primary antibodies was used overnight at 4 °C and incubated in properly diluted secondary antibody. Positive controls from Santa Cruz Biotech for each primary antibody were used for more accurate identification of specific antigens. WB results were normalized using GAPDH as loading control.Detection of antigen bands was displayed by WB chemiluminescent substrate(Thermo Fisher). The NIH Image program (Image J) was used for quantitative analysis of antigen bands. Representative data were derived from biological triplicates(mean+ standard error; SE).
Real-time PCR
RNA was extracted and purified from An3Ca and Ishikawa cell cultures to synthesize cDNA to determine the expression of NILCO componentsviaqPCR as previously described[8]. The following primers (Invitrogen, Carlsbad, CA) were used:Notch1 forward:5'-cactgtgggcgggtcc-3'and reverse:5'-gttgtattggttcggcaccat-3'; Notch2 forward:5'-aatccctgactccagaacg-3' and reverse:5'-tggtagaccaagtctgtgatg-3'; Notch3 forward:5'-tgaccgtactggcgagact-3' and reverse:ccgcttggctgcatcag-3'; Notch4 forward:5'-tagggctccccagctctc3'and reverse:5'-ggcaggtgcccccatt-3'; JAG1 forward:5'-gactcatcagccgtgtctca-3' and reverse:5'-tggggaacactcacactcaa-3'; DLL4 forward:tgctgctggtggcacttt-3' and reverse:5'-cttgtgaggtgcctggtt-3'; survivin forward:5'-gcccagtgtttcttctgctt3' and reverse:5'-cctcccaaagtgctggtatt-3'; Hey2 forward:5'-aaaaagctgaaatattgcaaat-3' and reverse:5'-gtaccgcgcaacttctgtt-3'. GAPDH forward:5'-agggctgcttttaactctggt-3'and reverse:5'-ccccacttgattttggaggga-3'. qPCR conditions and relative expression values (R) were calculated as described previously[12].Representative data were derived from triplicates (mean + SE).
MTT cell proliferation assay
HEC-1A, Ishikawa, An3Ca, and KLE cells were seeded in 96-well plates (5 × 103cells per well). Cells were serum-starved for 24 h and incubated for additional 24 h in medium containing several leptin concentrations. Then, 10 µL of 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) (5 mg/mL) were added to each well. After 4 h of incubation at 37 °C, cells were lysed by addition of 50µL DMSO per well. Absorbance was measured at 570 nm using a microplate reader(Molecular Devices, CA)[9].
Cell cycle assay
Ishikawa and An3Ca cells were cultured in 6 well plates and starved in serum-free medium as described above. After starvation, cells were treated with leptin (1.2 nM)and IONP-LPrA2 (0.0036 pM) for 24 h. The cells were trypsinized, washed with 1×PBS, and resuspended in cold 100% ethanol. Next, EmCa cells were fixed with 100%ethanol, washed and incubated with 50 µL PI staining solution for 40 min at 37°C. The cells were centrifuged to remove the PI and resuspended in PBS. Then, cell cycle progression was analyzed using a Cellometer Vision CBA system (Nexcelom Biosciences, Lawrence, MA)[8,9].
Blockade of IL-1 and leptin signaling
Ishikawa and An3Ca cells were serum-starved as described above and incubated with basal medium containing 1.2 nM leptin, 0.1 mg/mL rabbit IL-1R tI antibody and 0.0036 pM IONP-LPrA2 for 24 h. Notch expression levels were analyzed by WB as previously described.
Small interfering RNA
Specific small interfering RNAs (SiRNA, Qiagen) were used to suppress the gene expression of Notch1, Notch3, and Notch4 in Ishikawa and An3Ca cells. Cells were seeded at a density of 1 × 105/mL in 12 well plates and cultured in DMEM medium containing 10% FBS and 1% penicillin and streptomycin solution until they were 60%confluent. Conditioned media were removed, and cells were cultured for 6 h in serum-free basal medium containing Notch1, Notch3, or Notch4 oligonucleotides (10 nM) and controls. SiRNAs were composed of four specific siRNA targets of 19-25 nucleotide length and negative controls (SiControl) included scramble SiRNAs(Qiagen). After SiRNA transfection, cells were cultured at 37°C for an additional 24 h in DMEM with 10% FBS and antibiotics. Cell lysates were obtained and analyzed for WB using specific antibodies to determine Notch1, Notch3, and Notch4 expression.
Cell invasion assay
Ishikawa and An3Ca cells (8 × 104) were transfected with siRNA for Notch1, Notch3 and Notch4 as described above. Cells were suspended in DMEM-serum free medium(basal medium) and added to the upper chamber of an insert coated with matrigel(6.4-mm diameter, 8-mm pore size; BD Biosciences). The inserts were placed in 24-well plates containing basal medium with or without leptin (1.2 nM). Invasion assays were carried out for 24 h. Then, the cells in the upper side of the insert were whipped out with a cotton swab and cells in the lower side of the insert were fixed with 3.7%formaldehyde and stained with hematoxylin. Six randomly selected fields (× 10 objective) were photographed, and the migrated cells were counted[10].
PTX cytotoxicity
To assess whether leptin is a survival factor for EmCa treated with chemotherapeutics, Ishikawa and An3Ca cell lines were treated with Paclitaxel (PTX).
CCK8 and annexin IV assays
First, to determine the dose-cytotoxic effects of PTX in EmCa cells, Ishikawa and An3Ca cells were cultured in 96 well plates and starved as described and with treated different concentrations of PTX (50-1000 nM) in medium containing leptin (1.2 nM)and IONP-LPrA2 (0.0036 pM) for 3 d. Then, cells were incubated with 10 µL per well of the reagent from Cell Counting Kit-8 (CCK8; Dojindo Molecular Technologies, Inc.)for 4 h at 37°C. Absorbance was measured at 450 nm using a microplate reader(Molecular Devices, CA) to determine cell viability.
Additionally, apoptotic and viable cells were determined using the Annexin V FITC/PI Assay (Nexcelom) after treatment. Ishikawa and An3Ca cells were cultured in 6 well plates until approximately 80% confluency. Cells were starved as described above. Then, cells were treated with PTX as described above for 3 d. After that, cells were treated with Annexin V-FITC and propidium iodide (PI) staining solution. The number of apoptotic, necrotic and live cells were measured using Cellometer Vision CBA System (Nexcelom Biosciences, Lawrence, MA).
Statistical analysis
ANOVA and studentt-test were used for data analysis. Representative results were performed in triplicate. Values forP< 0.05 were considered statistically significant based on the F-values and Tukey's multiple comparisons between group means as determined using SigmaPlot (Systat Software, Inc.). Mean + SE are indicated in the graphical analysis, based on replicates of densitometry analysis of WB, the percentage of cell cycle sub-phases, or percentage of proliferating cells as indicated in the figures.
RESULTS
Leptin induces Notch protein and mRNA expression in EmCa cells
Overall, leptin increased protein levels of Notch receptors, ligands and downstream targets in a dose dependent manner in EmCa cell lines (Figure 1). Remarkably, at least a two-fold increase in levels of Notch receptors, ligands and targets were seen after leptin treatment of poorly differentiated cell lines [An3Ca (Figure 1C) and KLE(Figure 1D)]. Notch1, Notch3, and Notch4 receptors, Notch ligands (JAG1 and DLL4)and Notch targeted molecules (survivin and Hey2) were also significantly increased but to lesser extent by leptin in more differentiated type I EmCa cells:Ishikawa(Figure 1A) and HEC-1A cells (Figure 1B). Similarly, leptin induced 1.2-2.4 and 2.0-5.5 fold mRNA expression of Notch receptors, ligands and targets in type I (Figure 2A)and type II EmCa cells (Figure 2B), respectively.
Leptin up-regulates IL-1R tI and OB-R expression in EmCa cells
In basal culture conditions, type II EmCa cell lines (Figure 3C and 3D) expressed significantly higher IL-1 (IL-1R tI) and leptin (OB-R) receptors than type I EmCa cells(Figure 3A and 3B). Leptin increased more the expression of OB-R and IL-1R tI in type II EmCa cells (2.7-3.7 fold) compared to type I EmCa cells (1.6-2.1 fold) (Figure 3).
Leptin induces cell cycle progression and proliferation of EmCa cells
Leptin significantly increased S-phase progression (Figure 4A) and proliferation(Figure 4B) of EmCa cells. However, leptin-induced S-phase progression was higher in type II EmCa cells (2.0-fold increase) compared to type I EmCa cells (1.5-fold increase) (Figure 4A). Moreover, type I EmCa cells were less responsive to leptininduced proliferation (2.0-2.7 fold) compared to type II EmCa cells (3.0-3.5 fold)(Figure 4B).
Inhibition of IL-1R tI decreases leptin-induced Notch expression in EmCa cells
Next, we assessed whether a functional crosstalk between leptin and IL-1 signaling could be involved in the regulation of Notch expression in EmCa cells. The blockade of IL-1 signaling by an anti-IL-1R tI antibody significantly reduced leptin-induced upregulation of Notch receptors and ligands in type I (Figure 5A) and type II EmCa cells(Figure 5B). To further assess the specificity of leptin's effects in EmCa, we determined whether the inhibition of leptin signaling via IONP-LPrA2 could affect Notch expression. IONP-LPrA2 abrogated leptin-induced effects on Notch expression in EmCa cells (Figure 5A and 5B). These results indicate that a functional crosstalk between leptin, IL-1 and Notch (NILCO) occurs in EmCa.
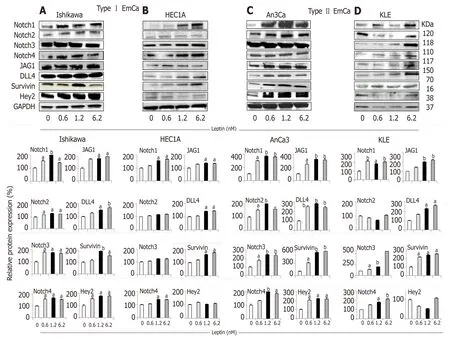
Figure 1 Leptin-induced Notch protein expression in endometrial cancer cells.
Notch signaling is involved in leptin-induced EmCa cancer cell invasion
We previously showed that the addition of DAPT (an inhibitor of gamma-secretase,the rate-limiting enzyme of Notch activation) reduced leptin-induced EmCa migration[16]. Similarly, leptin-induced migration of EmCa was abrogated by using anti IL-1R tI antibodies in EmCa cells[16]. To additionally examine the role of NILCO in EmCa, we determined whether leptin-induced cell migration is affected by the specific inhibition of Notch signaling via SiRNA. It was further accessed that leptin significantly induced invasion of EmCa cells that was more evident in the more aggressive An3Ca cells (type II EmCa). Notably, leptin-induced cell invasion was significant reduced by Notch siRNA knockdown (data not shown). However, no significant differences were found by silencing specific Notch receptors.
Leptin reduces PTX cytotoxicity of EmCa cells
PTX significantly reduced the viability of Ishikawa (PTX EC50 approximately 392 nM;Figure 6A) and An3Ca cells (PTX EC50 approximately 356 nM; Figure 6B).Remarkably, the addition of leptin significantly increased the number of live cells treated with PTX [type I EmCa (PTX-leptin EC50 approximately 458 nM; Figure 6A)and type II EmCa (PTX-leptin EC50 approximately 445 nM; Figure 6B)]. Interestingly,IONP-LPrA2 re-sensitized EmCa cells to the cytotoxic effects of PTX. Moreover, the addition of IONP-LPrA2 could allow for using a reduced quantity of PTX to achieve EC50 dose in both cell lines [type I EmCa (PTX-leptin-IONP-LPrA2 EC50 approximately 167 nM; Figure 6A) and type II EmCa (PTX-leptin-IONP-LPrA2 EC50 approximately 240nM; Figure 6B)]. These results suggest a potential for the use of IONP-LPrA2 as a chemotherapeutic adjuvant for EmCa.

Figure 2 Dose-response effects of leptin on the levels of Notch mRNA in endometrial cancer cells.
DISCUSSION
We earlier reported that the expression of NILCO molecules in EmCa tissues was associated with obesity. Furthermore, type II EmCa tissues expressed higher NILCO molecules, which suggests the signaling crosstalk is linked to the progression of the more aggressive EmCa phenotype[16]. However, whether leptin signaling could induce Notch expression and oncogenic changes in EmCa cells is poorly understood[20-22].Here we found that leptin is an inducer of Notch and a survival factor for EmCa cells.Moreover, NILCO was linked to proliferation, invasion and drug resistance (PTX),especially in type II EmCa.
Although, the categorical classification of EmCa in two major types (type I and II) it is still a point of discussion, endometrioid adenocarcinoma is known as type I EmCa,which is a more differentiated cancer that resembles normal endometrial morphology.Approximately 85% of all EmCa are type I and have better prognosis. Type I EmCa shows higher incidence and dependence of hormonal stimuli. In contrast, type II EmCa shows more aggressive phenotype, poor prognosis and is associated with high recurrences. Type II EmCa comprises less differentiated tumors showing lower incidence, but their growth and progression are independent of sex hormone stimuli.Additionally, there are other less abundant EmCa types that include the mixed mesenchymal Mullerian malignant tumor (MMMT) or carcinosarcoma, mucinous,clear cell, squamous cell, mixed and undifferentiated[2].
Obesity is a risk factor for EmCa, albeit through not very well-defined mechanisms[2,3]. High adiposity is accompanied with deregulated levels of many molecules(i.e., estrogen, leptin, leptin induced-molecules, Notch, cytokines and growth factors)that affect EmCa progression. Basal-like breast cancer and type II EmCa present similar genomic features and lack targeted therapies[2,14].
We have previously shown that the crosstalk between leptin, Notch and IL-1 can induce important oncogenic actions in breast cancer. Moreover, a functional leptin-Notch axis was also reported essential for pancreatic cancer progression and growth[5,8,9]. Aberrant Notch activation has been reported in many solid tumors.However, Notch signaling pathway can exert divergent impact on cancer tissues.Indeed, it is known that Notch signaling may be oncogenic or suppressive in different tumors[5]. Accumulating evidence suggest a role for Notch signaling in EmCa[2,15].However, the role of Notch in EmCa is still poorly understood. We hypothesize that NILCO could be a link between obesity and EmCa progression.

Figure 3 Leptin-induced IL-1R tI and OB-R protein expression in endometrial cancer cells.
Here, we present data supporting the notion that leptin can induce the expression of NILCO molecules in EmCa affecting cell proliferation, aggressiveness and chemoresistance. Present data suggest that leptin is an inducer of Notch [Notch receptors (Notch1-4), ligands (JAG1 and DLL4) and downstream effectors (survivin,Hey2)] and leptin (OB-R) and IL-1 (IL-1R tI) receptors in EmCa cells. Leptin showed higher impact on the poorly differentiated and more aggressive cell lines, An3Ca and KLE, which resemble type II EmCa. Remarkably, leptin treatment significantly increased mRNA expression of NILCO molecules in type II EmCa cells. Additionally,leptin also increased the expression of Notch1, Notch3, and Notch4 receptors in the more differentiated EmCa, HEC-1A and Ishikawa cells that resemble type I EmCa.
It was earlier reported that OB-R expression correlated with estrogen (ER) and progesterone receptor expression in EmCa[23]that suggests a potential crosstalk between leptin and estrogen signaling could occur in type I EmCa, which is more dependent on hormonal cues. We earlier reported that type II EmCa expresses higher levels of OB-R[16]. Present data show that OB-R and ER are co-expressed in EmCa.Additionally, we have previously found that leptin upregulates the IL-1 system in EmCa cells[11]. Here we extended these observations by showing that leptin greater increased OB-R and IL-1R tI expression in type II EmCa cells. Consequently, these cells were more responsive to leptin stimulus than type I EmCa cells in term of cell cycle progression, proliferation and invasion. Interestingly, as it was previously reported in breast[11], pancreatic[8,9], and some EmCa cells[16], leptin's effects on cell invasion were dependent of Notch signaling. Therefore, NILCO could be related to type II EmCa progression[5].
Surgery is the main treatment for EmCa. Among the more significant poor prognostic factors identified for EmCa treatment failure are initial stage II to IV, type 2 histology, positive cytology, and recurrence at multiple sites[24]. Adjuvant chemotherapy using single agent or combination is advised for metastatic, recurrent, or high-risk EmCa. PTX and combination therapies with platinium drugs or anthracyclines are commonly prescribed for EmCa aggressive disease[25]. Type II EmCa are independent of ER and PR signaling, very aggressive, have poor prognosis,and no targeted therapies[2]. Therefore, type II EmCa are mainly treated with chemotherapeutics, which eventually reduce their effectiveness due to the development of drug resistance. We hypothezise that leptin could be an important survival factor for EmCa treated with chemotherapeutics. Indeed, leptin's actions significantly reduced PTX cytotoxicity on EmCa cells. Moreover, inhibition of leptin signaling via IONP-LPrA2 re-sensitized EmCa to PTX's effects and allowed its dose reduction while achieving similar cytotoxicity on EmCa. These results encourage the notion for a potential use of leptin signaling inhibitors (i.e., IONP-LPrA2) as chemotherapeutic adjuvants for EmCa.
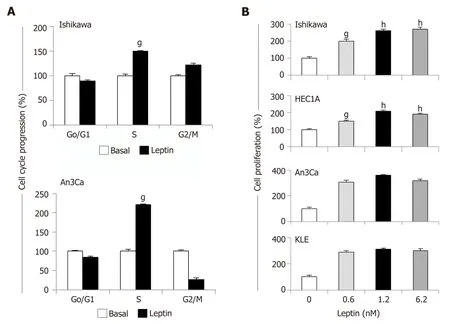
Figure 4 Leptin induces cell cycle progression and proliferation of endometrial cancer cells.
In conclusions, we report here for the first time that leptin is an inducer of Notch in EmCa. Leptin and Notch are part of a complex crosstalk (NILCO) that involves IL-1 signaling. Obesity (characterized by high levels of leptin) is a modifiable risk factor for EmCa that is linked to poor treatment outcome[2]. Thus, present data suggest that leptin through the induction of NILCO can increase EmCa aggressiveness and proliferation, and reduce PTX efficacy in type II EmCa cells, which showed higher responsiveness to leptin by overexpressing OB-R and Notch. Remarkably, the inhibition of leptin signaling re-sensitized EmCa cells to PTX. Since a strong association between central obesity and EmCa[3]has been found, present observations support the hypothesis that strategies leading to the inhibition of leptin signaling and NILCO in EmCa could be of paramount importance to design new adjuvant treatments to boost chemotherapy outcomes, and reduce chemotherapeutic dosage,especially in obese EmCa patients.

Figure 5 Inhibition of IL-1 R tI abrogates leptin induction of Notch in endometrial cancer cells.

Figure 6 Leptin increases survival of endometrial cancer cells treated with paclitaxel.
ARTICLE HIGHLIGHTS
Research background
The expression of Notch, Interleukin-1 (IL-1) and leptin outcome (NILCO) molecules (mRNAs and proteins) was previously detected in breast cancer and endometrial cancer (EmCa) from African-American and Chinese patients. Although, obesity status of Chinese patients was unknown, it looked like that NILCO was higher expressed in obese patients. However, NILCO molecules were expressed higher in type II EmCa, regardless of ethnic background or obesity status of patients. Leptin levels are high in obese patients that may suggest this adipokine is involved in the progression of the more aggressive EmCa phenotype (type II) and chemoresistance.
Research motivation
EmCa is the most frequent gynecological malignancy of the female reproductive tract and is the fourth most commonly diagnosed new cancer among women in the United States. Hormone nonresponsive breast cancer and type II EmCa have no targeted therapies, are mainly treated with chemotherapeutics and eventually develop drug resistance. Because leptin is a known inducer of NILCO in breast cancer and has been related to chemoresistance, it was hypothesized that comparable leptin's actions could occur in EmCa. The validation of this hypothesis may suggest that NILCO plays essential roles in tumor progression and chemoresistance, and thus,may represent a new EmCa target, particularly for type II EmCa.
Research objectives
To investigate whether leptin mediates the expression of NILCO signaling components, and impairs paclitaxel cytotoxic effects, and whether leptin's proliferative, invasion and chemoresistant actions are more prominent in type II EmCa cells.
Research methods
Two representative type I and type II (more aggressive and estrogen independent) EmCa cell lines were investigated for the potential leptin regulation of NILCO mRNA and proteins [Notch receptors, ligands and downstream effectors, and leptin (OB-R) and IL-1 (IL-1R tI) receptors]viaReal-time PCR and Western blot analysis. Leptin's proliferative and invasion effects were assessed by cytometric analysis (Cellometer Vision CBA system), and MTT and Matrigel-based invasion assays. NILCO inhibitors included nanoparticle-bound leptin peptide receptor antagonist-2 (IONP-LPrA2), anti-IL-1R tI antibody and Notch siRNA. The CCK8 assay was used to investigate leptin-mediated Paclitaxel drug resistance. Additionally, apoptotic and viable Paclitaxel-treated EmCa cells were determined by the Annexin V FITC/PI Assay (Nexcelom).
Research results
Leptin increased at least two-fold mRNA and protein levels of Notch receptors, ligands and downstream targets, and almost four-fold protein levels of OB-R and IL-1R tI in a dose dependent manner, mainly in type II EmCa cells. Leptin stimulated higher the progression of cell cycle, and the proliferation and invasion of type II EmCa cells, which were Notch-signaling dependent. The inhibition of IL-1R tI impaired the effects of leptin on Notch. Abrogation of Notch signaling via siRNA negatively affected leptin-induced EmCa invasiveness. Additionally,leptin acted as a survival factor for EmCa cells by significantly reducing the cytotoxic effects on Paclitaxel, which was more prominent in type II EmCa cells. The inhibition of OB-RviaIONPLPrA2 allowed the resensitization of EmCa cells to Paclitaxel. Thus, IONP-LPrA2 has a potential as a novel neo-adjuvant that may allow reducing Paclitaxel dosage and its undesirable side effects.
Research conclusions
For the first time, it was found that leptin is an inducer of Notch and targets in EmCa. Leptininduced NILCO could be specifically related to the progression, invasiveness and drug resistance of type II EmCa. Moreover, obesity could increase the progression of EmCa and the development of chemoresistance via leptin signaling and NILCO, which may be greater for type II EmCa. Leptin-induces NILCO, which could be a common signaling crosstalk that stimulates the progression and chemoresistance of several obesity-related cancers. Present data suggest that NILCO plays essential roles in tumor progression and chemoresistance, and thus, may represent a new EmCa target, particularly for type II EmCa.
Research perspectives
In vitro data suggest that EmCa requires leptin signaling and NILCO for proliferation and invasion, and to increase drug resistance and survival. Future research should investigate the role of NILCO in EmCa progression and chemoresistance using animal models. Spontaneous,syngeneic, xenograft and PDX EmCa-mouse models should be used to validate the hypothesis tested in the present paper.
 World Journal of Clinical Oncology2019年6期
World Journal of Clinical Oncology2019年6期
- World Journal of Clinical Oncology的其它文章
- Metastatic potential and prognostic significance of SOX2:A metaanalysis