
IL-6/gp130在类风湿性关节炎疾病中的作用:从机制到临床转化进展
2019-08-03 02:29吴锦雯张莹田吉来
药学研究 2019年7期
吴锦雯,张莹,田吉来
(1.南京中医药大学医学与生命科学学院药理学系,江苏 南京210023;2.南京大学医学院附属鼓楼医院风湿免疫科,江苏南京210008)
类风湿关节炎(Rheumatoid arthritis,RA)是一种慢性自身免疫性疾病,可导致关节、软骨和骨的持续性炎症和多重破坏,最终导致关节功能丧失,严重者可致残。据估计,RA在全世界的流行率为1%,并且其发病率正在急剧增加。RA的发病是一个多步骤的过程,最初始于关节外的血管前炎症阶段,随后是血管阶段,新生的血管增加。最后是滑膜异常增生和炎性细胞浸润,导致关节软骨和骨破坏,形成以缺氧环境和新血管生成为特征的关节损伤[1]。多种炎症细胞以及基质金属蛋白酶(matrix metalloproteinase,MMP)和肿瘤坏死因子(tumor necrosis factor,TNF)、白细胞介素-1(IL-1)、白细胞介素-6(IL-6)等炎症因子,在RA的发生发展中起主要作用[2]。
IL-6是一种多效细胞因子,参与慢性炎症的发生、自身抗体的产生、血管通透性的改变等过程。IL-6由基质细胞、单核细胞和淋巴细胞等产生,是RA急性期升高的重要炎症因子,和RA疾病严重程度成正相关[3],在某些极端情况下IL-6 水平可以从 1~5pg·mL-1急剧上升到几 μg·mL-1[4]。
在RA滑膜中,IL-6可在关节内募集白细胞,促进破骨细胞成熟和活化,抑制软骨细胞增殖,刺激滑膜细胞增殖,从而导致关节损伤。IL-6通过调节B细胞和Th17细胞分化,诱导和维持自身免疫过程。IL-6也可诱导细胞内黏附分子,参与血管生成。IL-6通过诱导RA中成纤维细胞样滑膜细胞(fibroblast-like synovial cells,FLSs)产生血管内皮生长因子(vascular endothelial growth factor,VEGF)调控血管通透性,从而促进炎症细胞向组织中的募集,加重损伤。IL-6在RA发病机制中的这些功能使IL-6成为RA治疗的显著靶点。
本文将总结以IL-6及其受体为靶标的RA治疗药物,讨论新型给药系统在RA治疗研究中的优势及进展,期望能为RA治疗药物的研发提供新的思路。
1 RA中的IL-6/gp130
IL-6受体(IL-6R)由 IL-6Rα(又称 CD126或 gp80)和IL-6Rβ(又称 CD130或 gp130)构成[5]。其中,IL-6Rα 是高亲和力的特异性配体结合链,分子量为80kDa,主要分布在肝细胞、中性粒细胞、巨噬细胞以及某些淋巴细胞等细胞表面。gp130的分子量为100kDa,糖基化后可达到130kDa,功能上作为信号转导链,几乎表达于所有细胞表面,包括心脏、肾脏、脾脏、肝脏、肺、胎盘和大脑等[5],且同时是 IL-11、IL-27、白血病抑制因子(leukemia inhibitory factor,LIF),抑癌蛋白M(oncostatin M,OSM)、睫状神经营养因子(ciliar yneurotrophic factor,CNTF)、心肌营养素(cardiotrophin,CT)-1、neuropoietin,humanin和心肌营养素样细胞因子(cardiotrophin like cytokine,CLC)的受体[6],在发育、造血、细胞存活和生长中起着重要作用。
目前发现,IL-6可通过3种模式与其受体结合,分别是经典信号途径(classics ignaling)、转移信号途径(或称反式信号途径,trans signaling)和聚类信号途径(cluster signaling,或trans presentation)[3](见图1)。IL-6与受体不同的作用模式主要取决于受体的类型。IL-6与膜结合型 IL-6Rα(membrane boundIL-6receptor,mbIL-6Rα)结合,进而招募gp130,形成三元六聚体复合物,介导经典信号途径。生物机体通过IL-6的经典信号途径,实现抗炎和保护的功能[7]。mbIL-6Rα可经剪切(splicing)或脱落(shedding)机制成为可溶型IL-6Rα(sIL-6Rα)。sIL-6Rα对 IL-6的亲和力与mbIL-6Rα相当。IL-6/sIL-6Rα复合物的形成不仅可以保护IL-6,维持信号活性并延长其循环半衰期,而且还可以作为一种激动剂,通过gp130介导转移信号途径,直接激活细胞。gp130在人体内广泛表达,因此,从理论上讲,IL-6/sIL-6Rα复合物可以刺激体内大多数细胞,即使在不表达IL-6Rα的细胞上也能结合和激活 gp130[8]。然而,可溶型gp130(sgp130)对这种转移信号有很强的调控作用,sgp130与IL-6/sIL-6Rα复合物结合,抑制IL-6/sIL-6Rα复合物与gp130的结合。因此sgp130是IL-6转移信号的天然抑制剂[4]。2017年,Heink等[9]首次报道了 DC 中存在 IL-6的聚类信号模式。IL-6在DC胞内与IL-6Rα结合后,被转运至细胞膜,膜上IL-6/IL-6Rα复合物可与靶细胞gp130结合并激活靶细胞。虽然sgp130可以干扰IL-6转移信号途径,但它不会影响聚类信号传导。聚类信号传导模式可以更快更稳的激活下游因子。
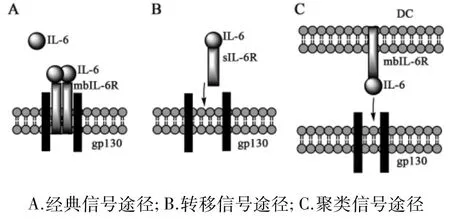
图1 IL-6介导的3种细胞信号转导途径
在经典信号途径中,IL-6先与细胞膜上的IL-6Rα(mIL-6Rα)结合,形成的 IL-6/IL-6Rα复合体再招募 gp130,形成IL-6/IL-6Rα/gp130六聚体。在转移信号途径中,IL-6与体液中可溶型IL-6Rα(sIL-6Rα)结合,形成的IL-6/IL-6Rα复合体,既可以与膜上的gp130结合,传递细胞信号,也可以与体液中的可溶型gp130(sgp130)结合而失活。在聚类信号途径中,IL-6与IL-6Rα在DC细胞内结合,形成IL-6/IL-6Rα复合物,并被运输至细胞膜上表达,靶细胞通过其自身的gp130接收和相应这一信号。
2 IL-6/gp130介导的信号转导
IL-6 通过JAK-STAT( Janus kinase-signal transducer and activator of transcription) 、ERK-MAPK( extracellular - signal - regulated kinase-mitogen - activated protein kinase)和PI3K - AKT( phosphoinositide 3-kinase-protein kinase B) 3种途径参与细胞的信号转导[10]。
在 JAK-STAT 途径中[6],IL-6/IL-6Rα/gp130 的六聚体,使gp130胞内近端的酪氨酸残基磷酸化和胞内的受体相关激酶(JAK1、JAK2和TYK2)被激活,STAT分子借SH2结构域接近JAK激酶而被磷酸化、活化,形成二聚体(包括同源二聚体STAT1/STAT1或STAT3/STAT3,异源二聚体STAT1/STAT3)移向核内,调节急性期蛋白基因的表达和开放。细胞因子信号转导抑制物(suppressor of cytokine signaling,SOCS)是JAK/STAT通路的靶基因之一。SOCS抑制JAK活性,从而负调控信号,提示该信号通路存在一种自我调节机制。骨髓基质细胞产生的IL-6可经JAK-STAT途径诱导核因子κB受体活化因子配体(receptor activator of the nuclear factorκB ligand,RANKL)激活,这是破骨细胞的分化、激活和骨吸收至关重要的环节。
ERK-MAPK途径可诱导滑膜细胞产生MMP,在IL-6刺激下,SHP-2被招募到gp130磷酸化的Tyr759残基中,随后被JAK磷酸化,然后与生长因子受体结合蛋白2(Grb2)相互作用,而Grb2与Ras的GDP/GTP交换器SOS有着内在的联系,使GDP转化为Ras-GTP并结合于膜上,激活Raf-ERK-MAPK级联,进而催化AP-1、NF-IL-6和TCF等核转录因子的磷酸化和活化,从而调节相关基因的表达和开放。
IL-6激活的第三条通路是PI3K/AKT通路,因为JAK可以磷酸化激活PI3K的酶,这种酶可将磷脂酰肌醇-4,5-二磷酸盐(PIP2)磷酸化为磷脂酰肌醇-3,4,5-三磷脂酰肌醇(PIP3)。PIP3反过来磷酸化和激活招募到质膜上的丝氨酸/苏氨酸激酶PkB/Akt,活化的Akt磷酸化几个下游靶点从而上调细胞分子表达水平(如图2所示)。
IL-6的目的基因包括survivin(BIRC5)、X染色体连锁的凋亡抑制蛋白(X linked inhibitor of apoptosis,XIAP)、Bcl-2、Bcl-XL、mcl1等生存相关蛋白、细胞增殖过程中涉及的细胞周期蛋白cyclinD1、MYC等蛋白和缺氧诱导因子(HIF)-1α、VEGF、碱性成纤维细胞生长因子(basic fibroblast growth factor,bFGF)、MMP-2、MMP-9 等促血管生成因子[11]。
此外,IL-6与多种促进其致瘤活性的途径发生相互作用,特别是环氧合酶-2(Cox-2)、Wnt、转化生长因子(TGF)-β和NFκB。IL-6可以刺激Cox-2在成骨细胞、破骨细胞中的表达,以及前列腺素E2(PGE2)的产生。PGE2通过增加成骨细胞RANKL的表达和破骨细胞RANK的表达,发挥破骨细胞活化的中介作用。此外,IL-6诱导成骨细胞中PGE2受体、EP2、EP4的表达,触发正反馈回路。随后PGE2刺激IL-6的表达,产生一系列增加骨溶解的信号。Wnt信号通路抑制因子Dickkopf(DKK)-1由乳腺癌细胞、骨髓瘤等多种转移性癌细胞表达,抑制骨形成并伴有骨溶解性转移。IL-6通过刺激骨髓瘤细胞中DKK-1的产生,阻止成骨细胞祖细胞分化为成熟成骨细胞,防止促进骨溶解。TGF-β和IL-6协同作用,增强骨降解。TGF-β上调成纤维细胞、成骨细胞、前列腺癌细胞等多种细胞类型中IL-6的表达,并刺激肿瘤细胞中PTHrP的产生。
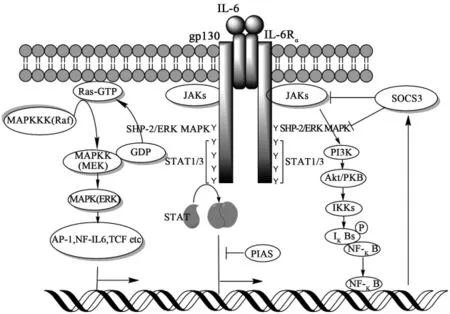
图2 IL-6/IL-6Rα/gp130六聚体通过3种通路激活靶细胞:JAK-STAT通路、ERK-MAPK通路和PI3K-AKT通路
在JAK-STAT中,IL-6/IL-6Rα/gp130六聚体使结合在gp130胞内部分的JAK被激活,并磷酸化gp130胞内部分的5个酪氨酸残基。磷酸化的近膜端酪氨酸触发ERK-MAPK通路。另外4个被磷酸化的酪氨酸残基会激活STAT1和/或STAT3,而STAT3则会磷酸化、二聚体化并转运到细胞核中作为转录因子参与靶基因的转录。SOCS3是gp130的负反馈调节因子,被STAT3转录激活,阻断JAK和ERK-MAPK通路。在ERK-MAPK通路中,SHP-2在IL-6刺激下被招募到gp130磷酸化的Tyr759残基中,随后被JAK磷酸化,激活SOS交换器,使GDP转化为Ras-GTP并结合于膜上,激活Raf-MEK-MAPK级联,进而催化AP-1、NF-IL-6等核转录因子的磷酸化和活化,从而激活和调节相关基因的表达。
3 靶向IL-6/gp130治疗RA的相关药物
IL-6既与关节炎症有关,也与许多关节外表现(如贫血、疲劳、心血管风险增加和骨丢失)有关。这推动了Tocilizumab(托珠单抗,罗氏Actemra,TCZ)的发展,在对TNF-17抑制剂和甲氨蝶呤(MTX)反应不足的患者进行了多项随机对照试验后,TCZ被批准用于治疗RA,成了临床上第一种人源化抗IL-6Rα单克隆抗体。TCZ的出现促进了以IL-6靶标途径的抗风湿病药物(disease-modifying antirheumatic drugs,DMARDs)的发展,包括靶向IL-6细胞因子的Sirukumab、Olokizumab和Clazakizumab等;靶向 IL-6Rα的 Sarilumab和靶向IL-6/IL-6Rα复合物的sgp130Fc。但其中大多数仍处于临床实验阶段[12],并且几乎都伴有严重的不良反应,如恶性感染、恶性肿瘤等。
3.1 以JAKs为靶点的药物 Tofacitinib(CP-690550)是一种新型的口服JAK抑制剂,主要抑制JAK3和/或JAK1杂二聚体的信号通路。它通过调节淋巴细胞功能中涉及的部分细胞因子信号通路来抑制某些免疫反应[13],在临床上常与MTX联用。
3.2 以IL-6为靶点的药物 Olokizumab(OKZ,CDP6038)是针对IL-6的人源化IgG4单克隆抗体,目前正在开发用于治疗RA。在Ⅰ期和Ⅱa(接受MTX治疗)临床试验中,OKZ在静脉和皮下给药后耐受性良好,平均血浆半衰期约31d,生物利用度84%~93%,无明显的抗体介导清除。在RA患者皮下单次给药12周后,OKZ还能显著降低游离IL-6水平和抑制C反应蛋白(C-reactiveprotein,CRP)。此外,在一项对先前抗TNF治疗失败的RA患者进行的剂量范围、双盲研究中显示,OKZ短期治疗既安全又有效,与 TCZ相当。Sirukumab(SRK,CNTO136)可以高亲和地选择性结合IL-6的人单克隆抗体,抑制IL-6经典和转移信号转导途径,对于MTX不耐受的患者,SRK能一定程度上减轻症状。Siltuximab(CNTO328)是一种IL-6靶向的人鼠嵌合单克隆抗体,可变区来源于鼠抗IL-6抗体CLB8,恒定区来源于人类IgG1κ分子,分子量约为145000,以高亲和力特异性结合和中和人IL-6,抑制IL-6与IL-6Rα的结合,从而阻断IL-6/gp130信号转导通路,进而抑制炎症和抗肿瘤活性。Siltuximab半衰期长(近2周),免疫原性不明显。因此在临床上比Elsilimomab(BE-8)更有益。此外,还有Clazakizumab、mAb1339(OP-R003)、PF-04236921、MEDI5117、C326(AMG-220)等在研靶向IL-6的生物药物。
3.3 以IL-6Rα为靶点的药物 TCZ是一种人源化的抗IL-6Rα单克隆抗体[14]。在先前临床研究的基础上,它于2008年在日本被批准为抗风湿药物,随后于2009年在欧洲和2010年在美国被批准。在RA活跃且对DMARD反应不足的患者中,使用TCZ抑制IL-6受体可减轻关节肿胀和压痛,改善生理功能,并降低影像学进展速度。TCZ存在潜在的免疫效应包括诱导或扩增B-调节细胞,减少促炎细胞因子和趋化因子基因的表达,促进滑膜液中与愈合相关基因的表达[15],在调节关节炎症以及风湿性关节炎的关节外表现和并发症(如疲劳、贫血、骨质疏松、抑郁、II型糖尿病和心血管风险增加等方面)中发挥着至关重要的作用[16]。
Sarilumab(SAR153191/REGN88)是一个全人源化抗IL-6Rα单克隆抗体,可特异性结合mIL-6Rα和sIL-6Rα,从而阻断IL-6介导的经典和转移炎症信号级联,并没有证据表明补体依赖或抗体依赖介导的细胞毒性。Sarilumab已经在临床前研究中被证明能够以剂量依赖的方式抑制IL-6信号转导[17]。
3.4 以 IL-6/IL-6Rα为靶点的药物 Sgp130Fc(FE 999301)由gp130的整个细胞外部分与人IgG1抗体的Fc部分融合,能与IL-6/sIL-6Rα复合物结合,与单独的IL-6或IL-6Rα无亲和力,是目前唯一一种特异性抑制IL-6转移信号的治疗剂。
3.5 以IL-6/gp130为靶点 现有诸多小分子gp130抑制剂处于研发阶段,如MadindolineA、SC144等,它们被广泛地应用于抑制 IL-6/gp130发挥抗癌作用[18]。Li等[19]利用计算机多配体同时拼接技术(multiple ligand simultaneous docking,MLSD)发现,雷洛昔芬(raloxifene)和巴多昔芬(bazedoxifene)均能够和gp130的D1区域进行有效结合,巴多昔芬的吲哚部分和七元氮杂环分别能模拟IL-6基团的Trp157和Leu57,进而可以实现拮抗IL-6的目的,并随后在人胰腺癌、人横纹肌肉瘤、及肝癌等细胞和动物水平的实验中得到验证[20-23],但是巴多昔芬和雷洛昔芬在 RA中的应用尚无报道。
综上,由于一些药物同时是mIL-6Rα和sIL-6Rα的拮抗剂,可同时抑制IL-6介导的经典和转移信号通路,因此带来不同程度的毒副作用和不良反应,如TCZ,其不良反应发生率较高(约为27.3%),易引发感染、肺炎、内脏炎症、带状疱疹、非典型性分枝杆菌感染、憩室炎、肺结核等[14]。
与单克隆抗体等生物制剂相比,小分子在给药途径、靶向选择性和特异性、安全性和有效性以及开发路径和总成本等方面存在诸多优势,例如:小分子药物成本低廉;给药方式通常为口服,患者依从性高;具有直接靶向胞内信号通路的能力。小分子IL-6/gp130抑制剂应用于RA的治疗被寄予厚望。但需注意的是,小分子药物的靶向性和特异性较单克隆抗体药物差,虽然激酶抑制剂不需要绝对的特异性即可应用于临床,但仍需警惕不良反应的发生[24]。
4 RA治疗的靶向给药系统
由于IL-6R分布广,IL-6的生物效应多样,全身给药易产生不同程度的不良反应。为了改善生物药物给药存在的不足,以及小分子药物的靶向能力不强的缺陷,为了减少传统药物所存在严重的副作用,如骨质疏松、肌肉萎缩、免疫功能受损等,减少IL-6/gp130分子靶向药的脱靶效应(offtargeteffects)风险,用于RA治疗的靶向给药或局部给药系统成为研究关注的重点。在细胞层面,载体设计可靶向到FLSs、VECs、炎症相关的巨噬细胞及T细胞等。
4.1 靶向给药的载体 纳米载体药物具有靶向输运和治疗的潜能。用于RA治疗的纳米载体研究最多的是脂质体、固态脂质纳米粒、聚合物纳米粒、树枝状大分子和金属纳米粒等[1,25]。
脂质体(liposome)是由脂质双层膜组成的球状纳米颗粒,药物可装入内部为水相或脂膜中。脂质体的理化性质包括渗透性、电荷密度和空间位阻,影响药物传输。脂质体可以改善关节腔内药物的疗效,减少药物的副作用[26]。在脂质体中加入乙醇或边缘活化剂可以增加变形能力。柔性脂质体可以通过其挤压细胞间隙的能力增强皮肤渗透性[27]。固体脂质纳米粒(solid lipid nanoparticles,SLNs)通常是由天然或合成的长链脂肪酸、脂肪醇或甘油三酯制备而成[28],在物理稳定性、药物的长期作用和高生物相容性方面优于其他胶体系统。在优化的SLN分散体组成下,物理稳定性可达3年以上。然而,固体脂质完美的脂质晶体限制了药物的溶解度,这就导致了SLNs系统中药物排出、载药量不足和纳米粒浓度较低的现象(1%~30%)[29]。脂质类载体往往约需1周的较短给药间隔,需要重复给药以维持治疗效果。
纳米金(gold nanoparticles,AuNPs)、纳米氧化铁(iron oxide nanoparticles)、纳米银(silver nanoparticles)等表面可合成或修饰几个官能团,进而能与受体、抗原等识别和结合,可结合自身属性用于药物的靶向诊疗。例如,AuNPs能够结合VEGF,在RA治疗中发挥具有抗血管生成作用;AuNPs通过清除ROS,抑制RANKL诱导的破骨细胞形成[30]。芦丁稳定的纳米银可通过抑制TNF-α和IL-6,发挥RA治疗中的抗炎作用[25]。纳米氧化铁,可以标记RA干细胞治疗中待移植的干细胞,可以标记免疫细胞,监测其浸润情况[31]。此外纳米氧化铁可以调节巨噬细胞极性的转变[32-33],影响炎症进展。
改善的渗透和滞留效应(enhanced permeability and retention,EPR)原理依然适用于 RA的被动靶向纳米载体治疗[34]。炎症部位的血管内皮细胞间有高达700nm的间隙生成,合适粒径的纳米载体可进入并滞留于滑膜组织,在靶点释放药物。纳米载体粒径的大小依然是被动靶向的考虑因素之一。
4.2 主动靶向修饰 透明质酸(hyaluronic acid,HA)通常存在于关节、软骨、眼和皮肤组织周围的滑液中,是一种高分子糖胺聚糖。透明质酸的治疗具有持久、止痛、保湿、润滑和皮肤充盈的作用,提高了组织的水化、弹性和耐久性。Choi等证明,在伤口愈合过程中,HA与IL-6具有协同效应,联合应用可显著提高划伤创面愈合实验中的细胞迁移率[35]。在慢性膀胱炎大鼠模型中,膀胱内注射HA可降低IL-6水平,从而降低炎症程度[36]。
另一方面,HA的受体CD44在RA发炎关节的滑膜淋巴细胞、巨噬细胞和成纤维细胞表面高表达,因此成为研究治疗RA主动靶向材料的首选。Gouveia等[37]发展了HA共轭修饰的HA-DPPE(1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine)pH敏感型脂质体用于RA的治疗,细胞摄取明显增加。Alam等[38]发展的HA修饰的磷酸钙纳米粒可以改善药物在胶原诱导关节炎的小鼠(CIAmice)中关节炎爪的生物分布。Zhou等[39]发展了载泼尼松龙的HA-SLNs,静脉给药后体内循环时间长,更易集中在骨和软骨组织,减少了关节肿胀、骨侵蚀和血清中的炎性细胞因子水平。采用TCZ修饰的HA-纳米金(HA-AuNP/TCZ),在CIA鼠中的治疗效果优于TCZ和HA-AuNP复合物,可能由于HA-AuNP/TCZ具有长效和对VEGF和IL-6R双重靶向的协同作用[30]。
由于激活的巨噬细胞表面高表达叶酸受体-β,成为区别于静息巨噬细胞和其他白细胞的特异性分子[40],故常用叶酸做靶头分子靶向RA的滑膜巨噬细胞[41]。由于RA炎症部位的VECs高表达黏附分子(如 αvβ3-整合素、E-选择素),使得 VECs成为主动靶向递送的潜在靶标[34]。利用RGD(Arg-Gly-Asp)靶向滑膜血管整合素,成为RA研究中的较成熟的靶向技术。
为了提高纳米载体的生物相容性,仿生血小板和RA之间的内在关系,血小板膜应用于包裹PLGA纳米粒(plateletmimetic nanoparticles,PNPs),用于RA的靶向药物递送,显著改善对P-选择素和糖蛋白VI的识别,表明PNPs可类似天然血小板,通过多种机制有效靶向RA组织[42]。受巨噬细胞固有的炎症靶向能力启发,采用细胞松弛素B减弱细胞骨架和巨噬细胞膜之间的相互作用,刺激巨噬细胞分泌荷膜囊泡(macrophage-derived microvesicle,MMV),其膜蛋白与巨噬细胞相似,用于包裹PLGA纳米粒,能够模拟巨噬细胞,成为RA靶向和治疗的有效仿生载体[43]。
4.3 局部给药 微针(microneedles)和离子透皮给药(iontophoresis)技术常用于经皮递药,它们可以通过改变皮肤表面结构,暂时突破皮肤,增强药物渗透性[44]。微针的设计目的是在皮肤表面建立机械通道,而不触及真皮层,以加强药物在皮肤屏障上的运输,几乎没有疼痛。离子透皮给药是一种非侵入性的透皮给药方法,其原理是利用低强度电流传递带电分子,药物可以更快地释放到皮肤中,具有释放大分子的能力,能更好地控制给药剂量等[45]。
关节内(Intra-articular,IA)给药存在着药物易被清除的缺陷,药效往往因此降低[46]。为了增加药物在IA的滞留,调控药物释放,提高药物生物利用度,科研人员研发了一些新的IA给药系统。ReumSon等[47]将包裹地塞米松的PLGA微囊分散在含有MTX的HA中,制备了荷载双药的水凝胶系统用以IA注射,实现了两种药物的先后释放和快慢释放,符合疾病治疗的需要,从而增强了RA的修复。值得注意的是,HA的半衰期较短(在组织中只有1~2d),未经修饰的HA存在降解速度快、机械性能差和清除速度快的缺陷[46]。Kim课题组继续发展了甲氧基聚乙二醇-b-聚ε-己内酯-ran-聚 L-乳酸[(methoxy)polyethylene glycol-b-poly( -caprolactone)-ran-poly(L-lacticacid),MC]二嵌段共聚物和MC支链末端羧基化的聚合物(MC-C),由于羧基的修饰MC-C呈现负电性,并根据阳离子或阴离子电解质药物对聚电解质材料产生不同吸引或排斥的作用,用于电负性的柳氮磺胺吡啶(sulfasalazine,Sul)和电正性的米诺环素(minocycline,Min)的凝胶制剂的调控释药[48]。静电作用的存在,影响了材料的胶凝时间,溶胶相变和药物释放。通过IA给予Min-MCC,改善了RA症状。Chiesa等[49]合成了可靶向表皮生长因子受体(epidermal growth factor receptor,EGFR)的 GE11多肽修饰的PLGA纳米粒,包裹地塞米松药物,然后将此分散壳聚糖基水凝胶中,开发了IA给药的制剂,该水凝胶能够在生理温度下15min内迅速从液态转变为凝胶态,延长了药物的局部释放时间。
5 展望
综上所述,IL-6作为一个相当有前景的治疗靶点,在临床上已多有运用,但是由于其抑制剂阻碍了正常的免疫应答,有很强烈的不良反应,并且此类制剂往往价格昂贵。纳米制剂等新型给药系统,可以发挥联合递药、诊疗一体、多药协同、局部施药和靶向可控递药等相结合的特点,有望成为RA可供选择的治疗手段之一。通过构建具有合适粒径和表面亲水性的纳米载体,经主动靶向材料修饰后,以局部给药的方式用于RA病区,进而可以发挥纳米药物在器官、组织、细胞多层次的逐级靶向和渗透的优势。期望未来能够研发出既可精准靶向RA病变区,又能选择性靶向IL-6/gp1303种模式的相关药物,降低不良反应的发生率,增加药物作用个时间,提高用药依顺性,从而改善RA患者的生活质量。
猜你喜欢
中风与神经疾病杂志(2022年9期)2022-10-19
中老年保健(2022年1期)2022-08-17
保健医苑(2022年5期)2022-06-10
昆明医科大学学报(2022年2期)2022-03-29
波谱学杂志(2022年1期)2022-03-15
昆明医科大学学报(2022年1期)2022-02-28
中老年保健(2021年3期)2021-12-03
昆明医科大学学报(2021年2期)2021-03-29
分析化学(2017年12期)2017-12-25
医学研究杂志(2015年7期)2015-06-22
