
Micromanaging autophagy with microRNAs to drive cancer metastasis
2019-07-29 04:54:22GracieWeeLingEngVenetiaJingTongKokJitKongCheong
Gracie Wee Ling Eng, Venetia Jing Tong Kok, Jit Kong Cheong
1Department of Biochemistry, Yong Loo Lin School of Medicine, National University of Singapore, Singapore 117597, Singapore.
2Medical Sciences Cluster, Yong Loo Lin School of Medicine, National University of Singapore, Singapore 117597, Singapore.
Abstract While we made great strides in the early detection of a handful of cancers, many other cancers are still detected at fairly later stages, thus hindering the deployment of effective surgical or therapeutic intervention to change their dismal clinical outcomes. The arduous journey of cancer cells from the primary tumor to colonize distant secondary organs or tissues begins with their ability to activate or deactivate various cellular processes at will, including the autophagy machinery. In this review, we discuss how circulatory cancer cells from primary tumors could selectively mobilize different subsets of microRNAs (miRNAs) to enable autophagic recycling of nutrients during their search for secondary sites to colonize and to disable such cell survival programs once they have successfully established at distant organs or tissues. We also discuss how this new miRNA-autophagy-metastasis axis can be targeted by the emerging RNA Medicine toolkit.
Keywords: Autophagy, miRNAs, cancer, metastasis
CANCER METASTASIS: KNOWING TOO LITTLE, TOO LATE
Metastasis remains one of the key reasons for the poor prognosis of cancer patients. The one-year survival of patients with stage 1 localized lung cancers is 87.3%, but plummets to merely 18.7% for stage 4 metastatic lung cancers[1]. Similar trend is observed for colorectal cancer, where one-year survival is 97.7% if detected at stage 1 and rapidly decline to 43.9% if detected at stage 4. These depressing statistics underscores our current knowledge gap in understanding the metastatic process, which impacts our ability to offer curative treatment options to cancer patients.
In the early stages of cancer, tumors are typically benign and remain confined within the normal boundaries of a tissue. As tumors grow and become malignant, however, they acquire the capacity to break through these boundaries and invade adjoining tissues. Notably, dissemination of tumor cells can occur relatively early in cancer progression[2]. During the process of metastasis, only a rare population of invasive cancer cells escape the primary tumor, survive the treacherous transit through the circulatory system,and subsequently establish secondary tumors in distant organs/tissues[3]. These invasive cancer cells,which exhibit extensive cytoskeletal remodeling, secrete proteases like extracellular-matrix-degrading metalloproteinases and cathepsins to drive their invasion and migration through the stroma, a network of supportive, connective tissue cells at the basement membrane[4,5]. Furthermore, the stroma can actively promote the initiation of metastasis by releasing transforming growth factor-β and other cell-signalling proteins to trigger cancer cells to undergo epithelial-to-mesenchymal transition (EMT) - a reversible phenotypic change in which cells lose intercellular adhesion and epithelial polarization and gain motility and invasiveness[6]. Transition into a mesenchymal cancer cell state allows for efficient intravasation of the lymphovascular system and acquisition of a stem-cell phenotype to promote survival during transit.These circulatory cancer cells can be arrested at distant organs/tissues, where they extravasate into the parenchyma of target organs to commence colonization. After extravasation, the invading cancer cells undergo mesenchymal-to-epithelial transition (MET) at the new settlement sites. The conduciveness of these niches for colonization can dictate the period of dormancy of these micrometastatic cancer cells. Long period of latency often exists between the development of a primary tumor and clinical manifestations of metastasis, indicating that metastatic colonization is a highly inefficient process in which most of these disseminated tumor cells (DTCs) die. To survive and establish macrometastases at distant organs/tissues, the minor subset of DTCs must overcome immune surveillance and other hosttissue defenses to achieve overt colonization at secondary sites[7]. Although breast-to-lung metastases have been shown to arise mainly from cells that have not undergone EMT in mouse models of breast cancer[8],it could be interpreted as rapid conversion into the epithelial cell state of DTCs, via MET, as soon as they arrive at supportive niches in the lung. Notably, the breast cancer cells that have undergone EMT are found to be resistant to chemotherapy and this could be attributed to their acquisition of a stem cell-like phenotype[9].
AUTOPHAGY: A CRUCIAL CANCER CELL SURVIVAL MECHANISM DURING METASTASIS
Autophagy is a cellular housekeeping mechanism which degrades and recycles bulks of cytoplasmic materials for the synthesis of essential cellular components[10]. Highly conserved from yeast to mammals,autophagy acts as an adaptive response to environmental stresses such as nutrient deprivation. The autophagy machinery has multiple distinct stages: initiation, nucleation, elongation, fusion and cargo recycling[10]. During stress, shifts in nutrient availability can be detected by proteins involved in energy homeostasis, such as the 5’ AMP-activated protein kinase (AMPK)[11]and the target of rapamycin complex 1(TORC1). The activation of AMPK and the corresponding inhibition of TORC1 will lead to the assembly of the Atg1/ULK complex, thereby kickstarting autophagy.
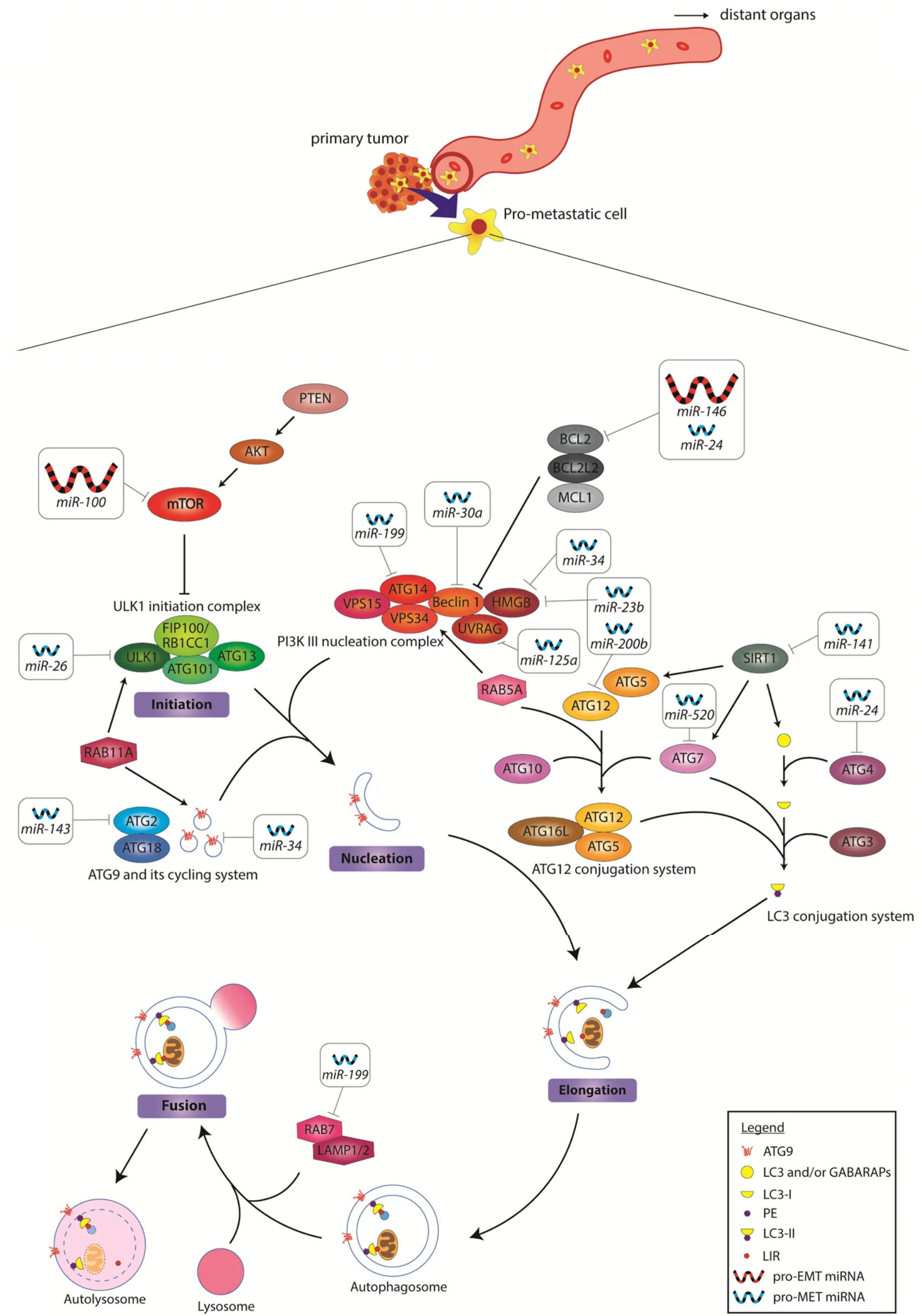
Figure 1. MiRNA regulation of autophagy during cancer invasion. The key components of macroautophagy is shown in this diagram.In the invading pro-metastatic cancer cell, autophagy is upregulated by gain of oncomiRs and loss of tumor-suppressive miRNAs. The oncomiRs target the inhibitors of autophagy to increase autophagic flux. The downregulated miRNAs on the other hand, are required for the activation of autophagy. Upregulated pro-EMT miRNAs are depicted as large red icons, while downregulated pro-MET miRNAs are depicted as small blue icons. The miRNA targets and mechanisms of actions are summarized in Table 1. EMT: epithelial-to-mesenchymal transition; oncomiRs: oncogenic miRNAs; MET: mesenchymal-to-epithelial transition
During autophagy, cytoplasmic cargoes are encapsulated by double-membrane vesicles known as autophagosomes and thereafter sent to the lysosomes to be degraded[12]. The intricate, multistep processes of autophagy are facilitated by numerous autophagy-related proteins (Atg). Detailed characterization of these proteins can be found in the review by Fenget al.[13]. At the initiation stage, the Atg1/ULK complex is assembled. This complex consists of Atg1/ULK, the regulatory subunit Atg13 and the scaffold complex Atg7-Atg31-Atg29 [Figure 1]. Importantly, the assembly of the Atg1/ULK complex is critical for the recruitment of downstream proteins to the phagophore assembly site. Next, Atg9 and its cycling system(Atg2, Atg18) are activated to facilitate membrane delivery to the expanding phagophore. This is followed by the assembly of the PI3K complex consisting of Vps34, Vps15, Vps30/Atg6, Atg14, and Atg38 [Figure 1],a process responsible for recruiting more proteins that are required for further phagophore expansion.Subsequently, two ubiquitin-like conjugation systems are activated for phagophore expansion, namely the Atg12 and the Atg8 conjugation systems. A detailed mechanism in which these conjugation systems function has been reported by Yinet al.[10]. The human homologs of Atg8 are categorized into two families- LC3 and GABARAP. Importantly, LC3 is employed during the phagophore elongation stage, whereas GABARAP proteins are only activated at a later stage of maturation. Upon induction of phagophore elongation, LC3 is cleaved at the C-terminal by ATG4B protease to form cytosolic LC3-I. This is followed by its subsequent conjugation and recruitment to the membrane to form LC3-II[13]. The level of LC3-II is often known to correlate with the number of autophagosomes due to its association with autophagosome maturation[14].
Fundamentally, LC3-II acts as an adaptor protein that selectively recruits cargoes to the autophagosome to be degraded[14]. Following cargo recruitment and closure of the autophagosome, the autophagosome directly fuses with a lysosome to form an autolysosome[12]. The cytosolic cargoes within the autolysosome are then broken down by lysosomal hydrolases, liberating building blocks for future synthesis of other macromolecules.
The importance of autophagy as a cellular homeostatic mechanism is underscored by the prevalence of autophagic defects in a myriad of diseases. Besides being a protein and organelle quality control mechanism that prevents the accumulation of aggregated and dysfunctional proteins, basal autophagy is also a highly regulated catabolic process that supports cellular metabolic and biosynthetic programs in response to nutrient deprivation and other forms of stress. In cancer pathogenesis, however, autophagy is hijacked by rapidly proliferating cancer cells as they adapt to the perturbations of the cancer microenvironment[15].
Autophagy is finely regulated in response to the specific energy demands or stress cues emitted throughout the different stages of metastasis - local invasion, intravasation, dissemination, extravasation and colonization at distant sites. Several studies suggested that autophagy is associated with EMT in cancer and activation of autophagy promotes metastasis[16]. This is corroborated by the high levels of autophagy detected in EMT-activated cancer cells, hence strongly supporting its role in promoting early metastasis[17].Additionally, cell detachment from the extracellular matrix typically has been shown to induce anoikis,a self-defense strategy that eliminates misplaced cells through apoptosis[18,19]. It then becomes crucial for cancer cells to evade anoikis so as to survive while traversing to distant organs/tissues. Importantly,several studies have postulated the pro-metastatic role of autophagy in overcoming anoikis[20-22]. Although the underlying mechanisms remain largely unknown, anoikis resistance conferred by autophagy has been shown to enhance the survival of cancer cells when they disseminate into the lymphovascular system,leading to their successful metastasis to distant organs.
Given that autophagy is a key cellular process that confers stress tolerance under adverse conditions, its activation in cancer cells that intravasate into the blood or lymphatic vessels enables them to withstand extreme metabolic stress induced by the changing environment[23]. To compensate for the sudden change in energy demands of the circulating DTCs, autophagy is activated to retain pools of functional mitochondria to sustain viability of the DTCs. In addition, autophagy-mediated degradation of proteins provides a sustainable source of nutrients for the DTCs to fuel ATP production and biosynthesis of other macromolecules that are essential for growth and survival[23,24].
Successful metastatic colonization is not only limited to the ability of DTCs to overcome stress that they are exposed to during circulation. Following extravasation from the blood vessel, the DTCs have been shown to switch from EMT to MET with concurrent inactivation of autophagy in supportive niches[25]. However,if the new environment is not permissive in situations such as cellular stress or a lack of available growth factors, the DTCs could exist in a dormant state, possibly by retaining elevated autophagic functions and delaying MET. Notably, autophagy inhibition in the neoadjuvant setting has also been shown to reduce pro-metastasis stromal cells at the premetastatic niche[26]. Future work could be performed to determine whether inactivation of autophagy is necessary to drive MET of invading tumor cells at distant organs/tissues, thus providing new impetus for the development of anti-metastasis autophagy inhibitors.
MIRNAS: NONCODING REGULATORS OF AUTOPHAGY AND CANCER METASTASIS
MicroRNAs (miRNAs) are endogenously expressed small non-coding RNAs of 19-25 nucleotides in length,which negatively regulate gene expression post-transcriptionally. They regulate multiple cellular processes including proliferation, differentiation and apoptosis. They have been implicated in a growing list of human diseases, such as diabetes and cancer[27].
In the canonical miRNA biogenesis pathway, the primary transcript (pri-miRNA) in the nucleus forms a characteristic stem loop structure, which is recognized and cleaved by the Drosha/DGCR8 heterodimer into pre-miRNA. The pre-miRNA hairpin loop is transported into the cytoplasm by Exportin-5, where its terminal loop is further cleaved by RNA endonuclease III Dicer to generate the mature duplex miRNA.Either strand can be processed into the final mature single-stranded miRNA, with the strand originating from the 5’end of the transcript known as 5p, while the strand originating from the 3’end is known as 3p. Association of the mature miRNA duplex with Argonaut proteins (Ago) forms the RNA-induced silencing complex (RISC), where the guide strand of ~22 nt remains in Ago while the passenger strand gets degraded[28,29]. Due to the short sequence length of miRNAs, and the fact that their partial complementary binding to multiple regions of target messenger RNAs (mRNAs) is sufficient to guide the RISC to its target genes, miRNAs are able to regulate the expression of a myriad of genes that play critical roles in controlling cellular processes, including autophagy and cancer metastasis.
AUTOPHAGY-REGULATING MIRNAS IN CANCER METASTASIS
Owing to the complexity and dynamic nature of autophagy, past literature that implicated a role of miRNAs in the regulation of autophagy have been highly discordant even in the same cell type. To reconcile and put things into perspective, we examined and discuss the opposing roles of miRNAregulated autophagy during the invasion phase versus the distant organ/tissue colonization phase of cancer metastasis.
At the onset of metastasis, cancer cells in the primary tumor first undergo EMT to enable the attached cancer cells to gain motile traits for invasion of the lymphovascular system. The role of autophagy in cancer cell motility and invasion was comprehensively reviewed by Mowerset al.[23]. Suffice to say that many reports have now demonstrated the elevation in autophagy in cancer cells drives tumor dissemination, and this may in part be regulated by miRNAs. miRNAs that have been shown to regulate autophagy and cell migration/invasion in the context of cancer have been summarized in Table 1.
Upregulation in oncogenic miRNAs (oncomiRs) is known to promote tumorigenesis[29]via modulation of gene expression of a variety of growth and survival pathways in cancer cells. For instance, elevated levels of miR-100 and miR-146 are found in cancer cells. Notably, these oncomiRs have been shown to be critical for the activation of pro-survival autophagy via suppression of the expression of autophagy inhibitory effectors like the mammalian target of rapamycin (mTOR) and Bcl2, respectively [Figure 1][30,32,33]. miR-100 and miR-146 have also been shown to be key EMT effectors that promote metastasis[31,34], with miR-100 proposed to act through HOXA inhibition[83], and miR-146 via Notch2 inhibition[84]. Overexpression ofthese oncomiRs provides several advantages to the invading cancer cells. Firstly, an increase in autophagy flux helps DTCs to resist anoikis following detachment from the primary tumor as well as to enable DTCs to gain access to nutrients to survive the arduous journey of traversing to distant organs/tissues.In addition, they concurrently change gene expression patterns in the cancer cells to allow these cells to become mesenchymal and thus more motile to invade distant sites.
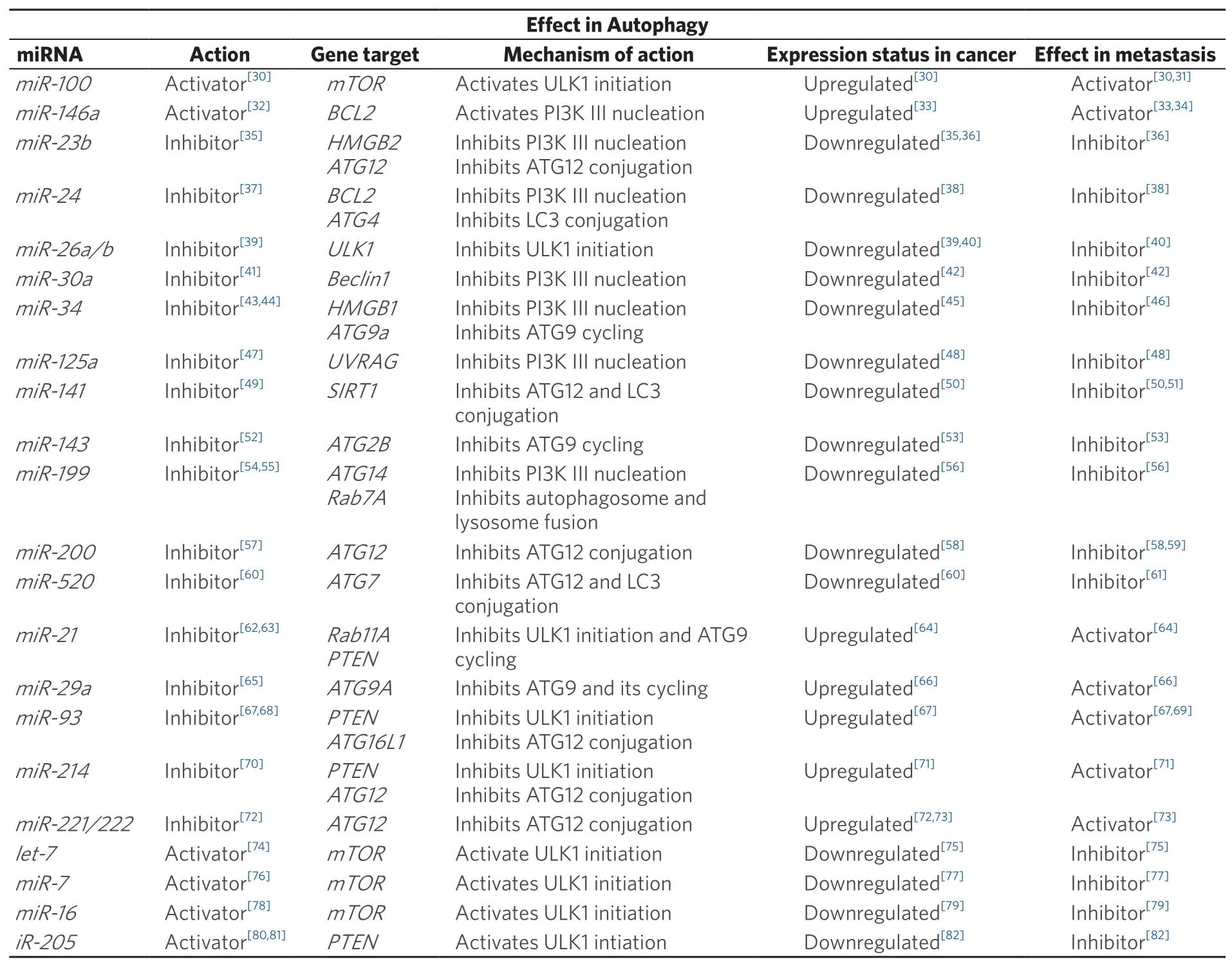
Table 1. MiRNAs and their effects on autophagy and metastasis
Apart from oncomiRs, the loss of tumor-suppressive miRNAs has also been shown to drive cell invasion and migration [Figure 1]. Several autophagy-inhibiting miRNAs, namely, miR-23b, miR-24,miR-26, miR-30a, miR-34, miR-125a, miR-141, miR-143, miR-199, miR-200 and miR-520 are found to be downregulated in cancer. Notably, these miRNAs are also known MET effectors[35-43,45-61,85,86]. As opposed to the primary role(s) of oncomiRs, we speculate that the loss of these miRNAs allows for higher autophagy flux and suppression of MET in the DTCs to help them adapt to unfavorable settlement sites.
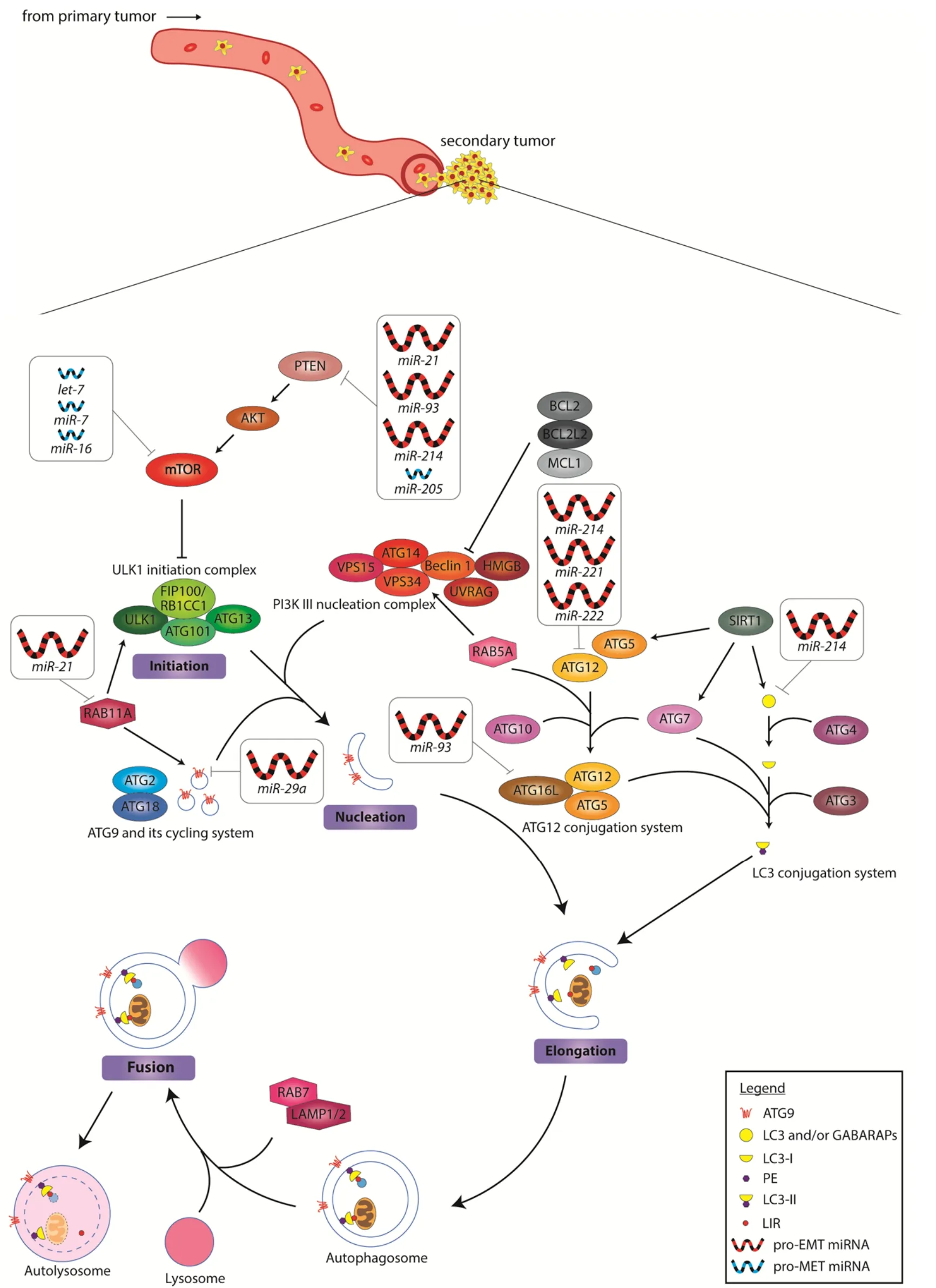
Figure 2. MiRNA inhibition of autophagy during secondary tumor formation. In the colonization process, autophagy is inhibited either by upregulation of anti-autophagy miRNAs, or the downregulation of the pro-autophagy miRNAs. This process promotes the reestablishment of an epithelial cell state for developing into macrometastasis. Upregulated pro-EMT miRNAs are depicted as large red icons, while downregulated pro-MET miRNAs are depicted as small blue icons. EMT: epithelial-to-mesenchymal transition; MET:mesenchymal-to-epithelial transition
Intriguingly, there exists a group of miRNAs that appear to block autophagy and yet promote EMT. These include oncogenic miR-21, miR-29a, miR-93, miR-214 and the miR-221/222 cluster that are highly expressed in cancer [Figure 2][62-73,87]. It seems counter-intuitive that successful distant organ/tissue invasion can be achieved for cancer cells without autophagy, but it remains plausible that the effects of these miRNAs might be context-dependent. For example, overexpression of miR-21 and miR-93 have been shown to induce metastasis by targeting PTEN[64,69], which in turn may promote the blockade of autophagy via activating the PI3K-Akt-mTOR signaling cascade. Since one miRNA can target many different genes, the biological context should also be taken into consideration in reconciling the opposing EMT-promoting and antiautophagy effects of miRNA-dependent loss of PTEN. Could miR-21 and miR-93 also regulate other targets concurrently to yield the phenotype observed? We also noted that several of these studies demonstrate induction of cell migration/invasion by overexpressing miR-21 or miR-93 in cancer cells grown in the tissue culture setting[64,67]. Unlike the circulating DTCs in a living organism, cancer cells culturedin vitroare never short of nutrient supply. It is perhaps not surprising that the need to trigger pro-survival autophagy in these cancer cells might be over-written by external culture conditionsin vitro. Alternatively,miRNAs like miR-21 and miR-93 may serve as sensors of favorable distant niches. As the DTCs extravasate at their destination sites, these miRNAs could respond to the new environment that provides nutrients to fuel the transition from micro- to macro-metastasis. Similarly, there exists tumor suppressive miRNAs that have been implicated in autophagy activation but yet inhibit cancer growth and metastasis. These miRNAs, including let-7, miR-7, miR-16 and miR-205, have been shown to be less abundant in tumors[74-82].Their absence in DTCs at micrometastasis sites might be crucial to cell survival, as autophagy genes will remain activated to ensure nutrient supply until conditions of the new environment turn favorable for macrometastasis.
PRECISION TARGETING OF MIRNA-AUTOPHAGY SIGNALING NODES BY SMALL MOLECULE AND RNA-BASED THERAPEUTICS
To date, cytotoxic chemotherapy remains the most widely used neoadjuvant or adjuvant treatment modality against cancers despite its association with a host of unpredictable side effects[88]. In addition, a recent study demonstrates that cytotoxic chemotherapeutics broadly used in neoadjuvant cancer therapy elicit tumour-derived extracellular vesicles with enhanced pro-metastatic capacity[89]. Hence, precise targeting of metastasis-enabling cellular processes, such as autophagy, is warranted and could synergize with chemotherapy to maximize therapeutic efficacy. For instance, small molecule inhibitor targeting of focal adhesion kinase (commonly known as FAK) activates the SRC kinase and thereby inhibits autophagy,which in turn, prevents the migration of SRC-driven metastatic tumor cells[90,91]. Given the complexity of signaling pathways driving metastasis, a systematic framework for the development of effective antimetastatic drugs has recently been proposed[92].
Of late, RNA-based therapies (antisense, siRNAs, aptamers, miRNA mimics/anti-miRs and synthetic mRNA) have shown great promise as powerful adjuncts to the drug developer’s existing toolbox of small molecules and biologics[93]. Notably, miRNA-based therapeutics gained much attention in translational medicine and are currently tested in various Phase I and II clinical trials. miRNA-based therapeutics is broadly categorized as miRNA mimics and inhibitors of miRNAs (also known as anti-miRs or antagomiRs). miRNA mimics, in the form of synthetic double-stranded small RNA molecules that match the corresponding miRNA sequence, are aimed at replenishing the lost miRNA expression in diseases.Most of the commercial miRNA mimics are often modified by methylation of the passenger strand to increase their stability. AntagomiRs, on the other hand, are antisense oligonucleotides that are singlestranded and can be chemically-modified (with locked nucleic acids or with 2'-O-methoxyethylribonucle oside) to increase affinity for their miRNA targets to block their functions[94]. To prevent degradation by RNases in biofluids or in the endocytic compartment of cells, these miRNA therapeutics are frequently stabilized by oligonucleotide chemistry (as mentioned above or by adding phosphorothioate-like groups)or encapsulated in delivery vehicles, such as poly(lactide-co-glycolide) particles, neutral lipid emulsions,synthetic polyethylenimine, cyclodextrin, chitosan,etc.,[95]. For instance, epidermal growth factor (EGFR)-targeted, EnGeneIC delivery vehicle-packaged miR-16 mimics have been tested in a Phase I clinical trial(NCT02369198) in which acceptable safety profile and on-target activity of these miR-16 mimics have been demonstrated in patients with malignant pleural mesothelioma[96]. In addition, two other clinical trials(NCT02862145 and NCT01829971), involving the use of miR-34a mimics to treat primary liver cancer, small cell lung cancer (SCLC), lymphoma, multiple myeloma, renal cell carcinoma and melanoma, also showed promising anti-cancer results[97]. Given that a subset of miRNAs mediate the crosstalk between autophagy and metastatic states of cancer cells, we postulate that the miRNA-autophagy-metastasis axis of cancer could be effectively targeted by the precisein vivodelivery of specific combinations of miRNA therapeutics(involving miRNAs depicted in Figures 1 and 2), which could improve the clinical trajectory of late stage cancers. Alternatively, blockade of the biogenesis of oncomiRs via small-molecule RNA ligands has also been reported to be a feasible anti-cancer approach[98]. These small molecules consist of high affinity RNA binding motives, including the aminoglycoside neomycin and different natural and artificial nucleobases,that target pri-miRNAs of oncogenic miR-372/miR-373 and inhibit Dicer-mediated maturation of these miRNAs to elicit anti-proliferative effects on gastric cancer cells.
Apart from miRNA therapeutics, siRNA therapeutics that target a number of driver oncogenes likec-Myc,PD-L1/2, mutantKRAS, PKN3, EphA2, LMP2/7, MECL1, APN401andPLK1have also been shown to exhibit on-target anti-cancer effects and elicit little or no overt toxicities in humans in a number of Phase I clinical trials (NCT00306904, NCT02314052, NCT02528682, NCT01676259, NCT01808638, NCT01591356,NCT00672542, NCT02166255 and NCT01437007). We envisage that delivery of siRNAs targeting genes that are important for autophagy and metastasis of cancer cells could also be a viable therapeutic route to combat late stage cancers.
CONCLUSION
Alterations in key cellular processes, including cell adhesive properties and autophagy, promote tumor cell dissemination and colonization at distant organs/tissues. These processes are mediated by a vast network of miRNAs that has the propensity to crosstalk with each other. Striking this Achilles heel of cancer precisely via specific combinations of miRNA therapeutics could dramatically improve the survival outcomes of cancer patients. Future work should be focused on tissue-specific targeting delivery vehicles, which carry miRNA therapeutics or other payloads, so as to avoid potential toxicities and off-target effects.
DECLARATIONS
Authors’ contributions
Manuscript writing: Eng GWL, Kok VJT, Cheong JK
Availability of data and materials
Not applicable.
Financial support and sponsorship
Eng GWL is supported by the Singapore Ministry of Education Postdoctoral Fellowship. Kok VJT is supported by the Singapore Ministry of Education (MOE) Academic Research Fund (AcRF) Tier 2 grant (MOE2016-T2-2-052). Cheong JK is supported by the Singapore MOE AcRF Tier 2 grant(MOE2016-T2-2-052) and a startup grant from the Medical Sciences Cluster of Yong Loo Lin School of Medicine, National University of Singapore.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2019.
 Journal of Cancer Metastasis and Treatment2019年9期
Journal of Cancer Metastasis and Treatment2019年9期
- Journal of Cancer Metastasis and Treatment的其它文章
- AUTHOR INSTRUCTIONS
- Liquid biopsy in lymphomas: a potential tool for refining diagnosis and disease monitoring
- Role of autophagy in therapeutic resistance of glioblastoma
- Membrane lipid binding molecules for the isolation of bona fide extraceIIuIar vesicIe types and associated biomarkers in Iiquid biopsy
- The immunological regulation of cancer cachexia and its therapeutic implications