
Mxr1磷酸化水平受Ptp调控机理的初步研究
2019-07-29 02:50侯成林杨艳坤陈嘉荔白仲虎
生物技术通报 2019年7期
侯成林 杨艳坤 陈嘉荔 白仲虎
(江南大学生物技术学院 碳水化合物与生物技术教育部重点实验室 工业生物技术教育部重点实验室 谷物发酵技术国家工程实验室,无锡 214122)
巴斯德毕赤酵母(Pichia pastoris,P. pastoris)是甲醇酵母。甲醇为单一碳源时,P. pastoris胞内的醇氧化酶 1(Alcohol oxidase 1,Aox1,EC1.1.3.13)能高效催化甲醇氧化成甲醛,同时 Aox1可达到细胞干重的30%[1-2]。利用高效的Aox1启动子(aox1promoter,Paox1)启动外源蛋白的表达,成功构建了巴斯德毕赤酵母表达系统,该表达系统是目前应用最广泛的重组蛋白表达系统之一[3-4]。然而P.pastoris甲醇的代谢存在碳源阻遏现象,即葡萄糖、甘油等碳源的存在可以阻遏甲醇的代谢,Paox1在甲醇为唯一碳源时被激活,在甘油存在时被抑制[5]。甘油代谢和甲醇代谢之间调控的分子机理尚不十分清晰,Paox1的激活和抑制是其研究的核心。
目前已发现多个调控Paox1的转录因子[6-7],其中甲醇表达调控因子1(Methanol expression regμLator 1,Mxr1,GenBank:ABD57365.1)是最重要的Paox1激活因子,Mxr1的缺失会导致P. pastoris无法利用甲醇[8-9]。研究表明,Mxr1是组成型表达,但是,在甲醇为唯一碳源的培养基中转录激活活性;在甘油培养基无活性[3,10]。同时发现Mxr1上存在磷酸化的Ser215位点,能够调控Mxr1转录激活活性[8]。说明Mxr1功能的发挥主要受磷酸化修饰调节。但是对Mxr1去磷酸化的酶一直尚未发现,本文在研究与Mxr1相互作用的蛋白中找到了使Mxr1去磷酸化的酶—蛋白质磷酸酪氨酸磷酸酶(Protein phosphotyrosine phosphatase,Ptp),并发现该酶对Mxr1的磷酸化水平的调节有明显的调控效果。蛋白质磷酸酪氨酸磷酸酶是一种比较保守的低分子量的磷酸酶[11-12],而蛋白的磷酸化和去磷酸化对蛋白活性的调节发挥重要作用[13],推测蛋白质磷酸酪氨酸磷酸酶调控Mxr1磷酸化,进而调控Mxr1活性。
1 材料和方法
1.1 材料
LA Taq DNA聚合酶、内切酶(EcoRI、XhoI、SacI)、T4 DNA 连接酶、卡那霉素、博来霉素、四环素,分光光度计(上海美诺达仪器有限公司),凝胶成像仪,蛋白质电泳仪(北京六一仪器),PCR仪(杭州朗基科学仪器有限公司)。
1.1.1 菌株和质粒 巴斯德毕赤酵母(P. pastoris)GS115、pGAPZB毕赤酵母表达载体、大肠杆菌E.coliDH5α,E. coliBL21,pSVT7大肠杆菌表达载体(表 1)。
1.1.2 培养基 LB培养基(酵母提取物5 g/L、胰蛋白胨10 g/L、NaCl 10 g/L、1×105Pa 灭菌20 min,固体培养基添加2%琼脂粉)、LLB培养基(酵母提取物5 g/L、胰蛋白胨10 g/L、NaCl 5 g/L、1×105Pa灭菌20 min,固体培养基添加2%琼脂粉)、BMY培养基(酵母提取物10 g/L、胰蛋白胨20 g/L、100mmol/L磷酸钾缓冲液,pH6.1,1×105Pa灭菌20 min,无菌酵母无氨基酸氮源YNB 13.4 g/L,无菌生物素0.4 mg/L,碳源按照实验要求后续添加)、YPD培养基(酵母提取物10 g/L、胰蛋白胨20 g/L、葡萄糖20 g/L、115℃灭菌30 min,固体培养基添加2%琼脂粉)。TB/SB培养基酵母提取物13 g/L、胰蛋白胨 24 g/L、 甘 油 10 g/L、KH2PO423.1 g/L、K2HPO412.55 g/L,1×105Pa灭菌 20 min。

表1 实验所用菌株及表型
1.1.3 引物 本研究所用到的引物(表2)由苏州金唯智生物科技有限公司合成。
1.2 方法
1.2.1 磷酸酶Ptp高表达载体的构建及宿主转化 以GS115基因组为模板,PCR扩增ptp片段,纯化,用KpnI,NotI分别双酶切片段和pGAPZB质粒,分化纯化回收,用T4连接酶链接片段和载体,得到ptp高表达载体PGAP-PTP,转化感受态E.coliDH5α,然后用含有博来霉素的LLB平板筛选和PCR菌落鉴定,并送苏州金唯智生物科技有限公司测序,保存测序正确的菌株。挑选重组子单菌落接种于YPD液体培养基培养24 h,提取基因组,用引物zecin-f,zecin-r扩增博来霉素基因。PCR产物用1%琼脂糖凝胶电泳,检测PGAP-PTP在P.pastoris染色体的整合情况。筛选到的重组子命名为PTP-GS115。
1.2.2 利用CRISPR/Cas9系统敲除磷酸酶ptp及构建Mxr1S215A定点突变菌株 利用本实验室构建好的基因编辑系统,设计3条sgRNA,电转cas9-GS115感受态,经过PCR,测序验证,在48碱基处成功敲除1个碱基,命名为ΔPTP-GS115。构建定点突变的上下游500 bp同源臂,通过融合PCR,形成上游同源臂定点突变下游同源臂序列,在突变位点上下200 bp左右设计3条sgRNA,构建sgRNA质粒,电转Cas9-GS115通过PCR验证敲掉的碱基数和定点突变。培养P.pastoris细胞、提取基因组及总蛋白质参照Invitrogen公司操作手册。

表2 实验所用的引物及sgRNA序列
1.2.3 荧光定量PCR检测Mxr1不同突变体下游基因表达量 将保存于YPD平板的突变菌株GS115和野生型GS115接种于YPD液体培养基进行过夜活化,统一OD600=0.5用无菌PBS洗3次,接种与含有1%甲醇为碳源的BMMY培养基,在30℃、230 r/min下培养24 h,36 h收集菌体,PBS清洗1次,提取RNA,反转成cDNA,荧光定量检测下游基因表达量。
1.2.4 Ptp诱导表达 通过NCBI数据库报道的ptp基因序列,由苏州金唯智生物科技有限公司对ptp序列进行密码子优化,6His-PTP连到pSVT7载体,转化感受态E.coliBL21,保存菌株。接种LB(15 μg/mL 四环素)的液体培养基,37℃,230 r/min培养过夜(14 h)。测定OD600,并按照初始0.1接种于TB/SB(含15 μg/mL 四环素)培养基中,30℃,230 r/min培养5 h,然后加入不同体积的IPTG进行诱导23℃,230 r/min培养16 h,收集菌体。超声波破碎细胞,提取总蛋白,磷酸酶的Ptp Western blot检测,总蛋白SDS-PAGE电泳后,使用电转仪将蛋白条带转移至PVDF膜上,TBST清洗3次,每次5 min。使用5%的脱脂奶粉封闭1 h,加入HRP标记的鼠抗His6抗体(1∶10 000),室温孵育1 h,TBST清洗3次,每次5 min,使用化学发光的方法显影,曝光时间 1 s[14-15]。
1.2.5 Ptp蛋白纯化 将pSVT7PTP-BL21(DE3)按照最优的发酵条件进行诱导表达,5 500 r/min收集菌体,加入与培养基等体积的上样缓冲液(20mmol/L NaH2PO4·2H2O、50 mmol/L NaCl,pH = 8.0)重悬。冰上预冷后进行超声破碎,破碎时间25 min,8 800 r/min离心收集上清。将上清用0.22 μm针头式滤器进行过滤,作为待纯化的样品。实验采用AKTA-purifier 蛋白纯化仪进行纯化,将滤出液经过Ni agarose蛋白纯化柱进行亲和层析,使用上样缓冲液平衡5CV以洗去未结合的杂蛋白质,再用洗脱缓冲液(20 mmol/L NaH2PO4·2H2O,50 mmol/L NaCl,250 mmol/L咪唑,pH8.0)洗脱,收集洗脱峰。将收集的蛋白质进行SDS-PAGE检测。将最优条件下发酵纯化的Ptp分装保存于-80℃[16]。
1.2.6 蛋白磷酸酶Ptp酶活验证 磷酸化多肽为底物,在蛋白金属磷酸酶(PMP)缓冲液。添加1mmol/L MnCl2,反应缓冲液,30℃温育30 min,反应体系如表3,ELISA检测反应产物,反应液包板4℃过夜,使用1%BSA的PBST室温封闭1 h,PBST洗板5遍,孵育相对应的磷酸化抗体,室温2 h,PBST洗板5次,室温孵育HRP标记的兔抗二抗1 h,PBST洗板5遍,加入TMB室温20 min,1 mol/L硫酸终止反应,多功能酶标仪检测结果。
1.2.7 Mxr1磷酸化检测 YPD过夜活化,OD=0.5转接BMMY和BMGY,24 h,按照 1.2.3的方法取样,统一OD600=4收集菌体,提取蛋白,经过Bradford法蛋白质定量,之后进行SDS-PAGE电泳后,使用电转仪将蛋白转移至PVDF膜上,TBST清洗3次,每次5 min。使用1%的BSA封闭1 h,P215磷酸化抗体(1∶2 000),4℃孵育过夜(14 h),TBST清洗3次,每次5 min,室温孵育HRP标记的兔抗二抗1 h使用化学发光的方法显影,曝光时间1 min。
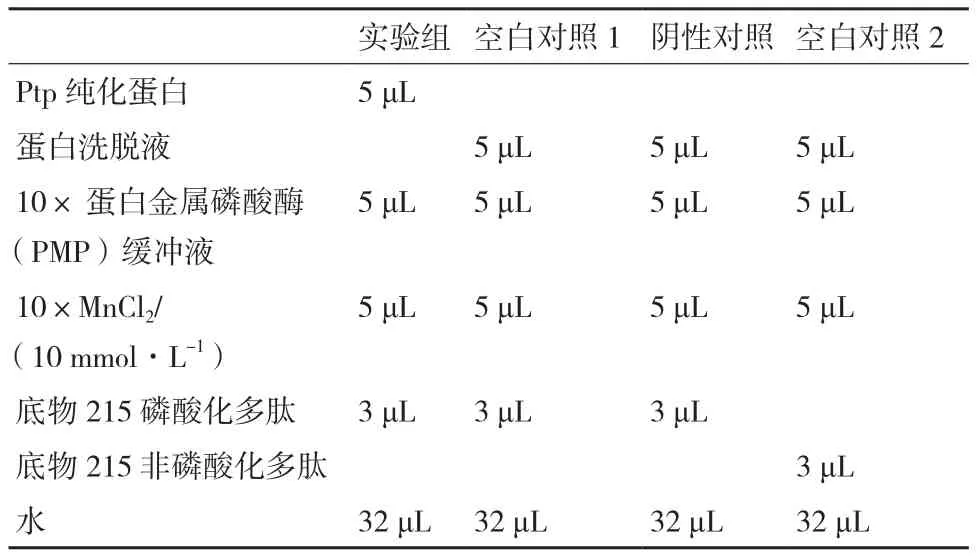
表3 ELISA不同实验中反应类型
2 结果
2.1 Ptp与Mxr1相互作用
为了寻找与Mxr1相互作用的蛋白,我们将Mxr1前400氨基酸与GST标签蛋白,表达纯化,将靶蛋白- GST融合蛋白亲和固化在谷胱甘肽亲和树脂上,用分别在甲醇和甘油培养的GS115蛋白溶液过柱,可从中捕获与之相互作用的“捕获蛋白”(目的蛋白),洗脱结合物后通过SDS-PAGE电泳,切胶,经过质谱分析,得到其中一个蛋白为Ptp(图1)。
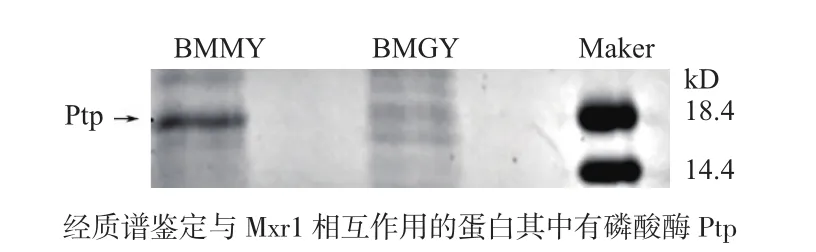
图1 Mxr1 PULL-DOWN 结果及Ptp条带
2.2 验证Ptp功能
为验证Ptp的去磷酸化功能,在大肠杆菌表达,诱导表达纯化脱盐后,经BCA法测得蛋白浓度是0.98 mg/L,以不同浓度pNPP为底物,相同体积酶反应30℃,10 min,1 mol/L硫酸钠终止反应,405 nm测吸光强度,测得Ptp米氏常数Km= 2.023 mmol/L,最适酶促反应温度40℃,最适pH4.5(图2)。
通过ELISA检测酶促反应结束后底物剩余量,将酶促反应的底物包板,每孔包被5 mg磷酸化抗原多肽,或非磷酸化抗原多肽,孵育磷酸化抗体(稀释比例 是 1∶2 000;1∶4 000;1∶8 000),通过加入纯化的Ptp蛋白和只加入蛋白洗脱液对比(图2-E),同样的反应条件,发现加入Ptp的样品几乎没有磷酸化抗原多肽剩余。说明纯化出的Ptp蛋白有去磷酸化的蛋白活性。
2.3 Mxr1磷酸化情况
按照1.2.4的方法取样,收集菌体,统一OD600=4收集菌体,提取蛋白,经过BCA法蛋白质定量,之后进行总蛋白SDS-PAGE电泳后,使用电转仪将蛋白转移至PVDF膜上,孵育P215磷酸化抗体(1∶2000)使用化学发光的方法显影,如图3野生型GS115在甘油培养基里没有发生磷酸化,在甲醇培养基里发生磷酸化,而Ptp高表达无论在甘油还是甲醇培养基都不发生磷酸化。
2.4 Mxr1定点突变和敲除在甲醇培养基中对与甲醇代谢相关基因的调控
敲除Mxr1后在BMMY培养基中Aox1几乎不转录,与甲醇代谢相关的酶的转录水平严重下降,将磷酸化位点定点突变后,与甲醇代谢相关酶的基因表达量也有所下降,说明Mxr1的磷酸化修饰调控下游基因的表达,尤其是与甲醇代谢相关的基因的表达(图4)。
3 讨论
Aox1启动子受甲醇诱导,但是其诱导机制到目前为止还没有完全揭示清楚,而Mxr1是毕赤酵母Aox1启动子的必要转录激活因子,为了研究甲醇的激活机理,先研究Mxr1激活机理,通过PULLDOWN发现与Mxr1相互作用的磷酸水解酶 Ptp,并对Ptp进行了高表达和敲除,发现在甲醇培养基中,高表达菌株Mxr1磷酸化程度远远高于敲除菌株。说明Ptp调控Mxr1磷酸化修饰。
本实验用Mxr1特定位点磷酸化多肽作为底物,发现Ptp识别Mxr1S215位丝氨酸位点的磷酸化,并将该位点磷酸基团去除。将Mxr1S215位丝氨酸突变成丙氨酸后,通过荧光定量PCR检测Mxr1野生型,敲除,定点突变菌株的下游基因表达量,发现与甲醇代谢相关的基因转录水平发生了改变,其中Aox1、Aox2、Das1、Das2定点突变菌株转录量相对于野生型的下降接近60%。以上说明Mxr1 S215位磷酸化修饰,影响与甲醇代谢相关基因的转录水平。蛋白的磷酸化修饰与蛋白的结构,亚细胞定位,功能息息相关[17-18]。Mxr1磷酸化修饰对其蛋白活性的影有可能是通过影响Mxr1空间结构,进而影响Mxr1蛋白活性。
在酿酒酵母中,Ptp作为一个高度保守的小分子蛋白磷酸酶,在AMPK途径发挥重要作用[12],根据小分子磷酸酶的特性推测其底物有可能不止Mxr1一个,需要通过PULL-DOWN找与Ptp相互作用的蛋白。寻找在甲醇代谢通路中Ptp底物,从这些底物磷酸化修饰入手,揭示甲醇诱导代谢途径所在的细胞信号通路。需要进一步验证Mxr1第215位丝氨酸磷酸化与其亚细胞定位是否相关,Mxr1磷酸化修饰是否控制Mxr1进出细胞核,还是调控Mxr1的活性,同时寻找Mxr1其他磷酸化位点,进而揭示Mxr1激活Aox1转录的分子机理,以及甲醇诱导Aox1转录机理。最终减少甲醇的用量,提高利用Aox1启动子表达外源蛋白生产工艺的优化及表达量。
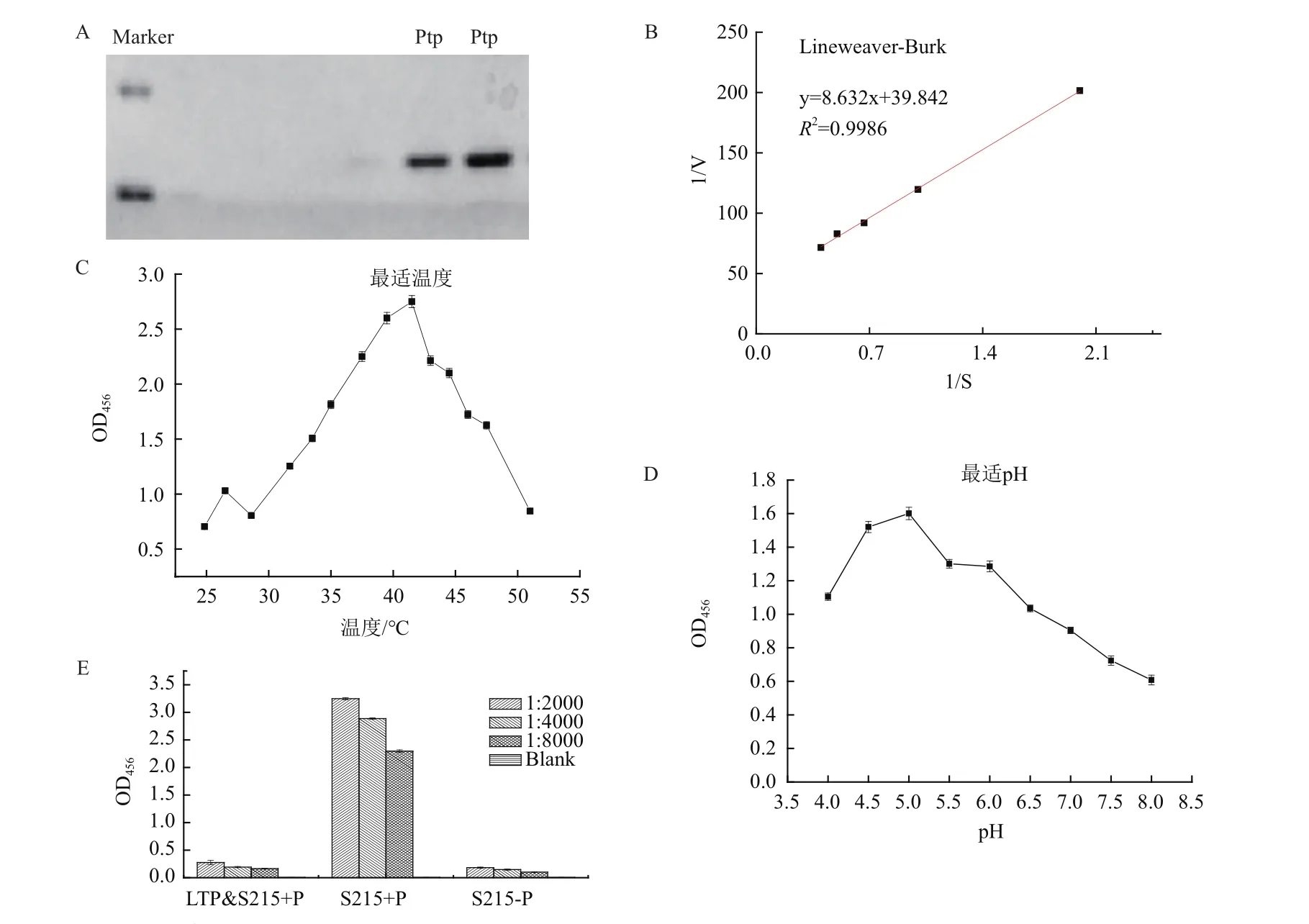
图2 Ptp蛋白外源表达纯化后的酶学性质研究

图3 在分别用甲醇和甘油为唯一碳源培养时,野生型,高表达和敲除菌株的磷酸化水平
4 结论
本研究发现与Mxr1相互作用的磷酸酶 Ptp,验证Ptp的功能是去除Mxr1S215位丝氨酸位点的磷酸基团。将Mxr1S215位丝氨酸突变成丙氨酸后,发现Aox1、Aox2、Das1、Das2定点突变菌株转录量相对于野生型的下降约60%。以上说明Mxr1 S215位磷酸化修饰,影响与甲醇代谢相关基因的转录。
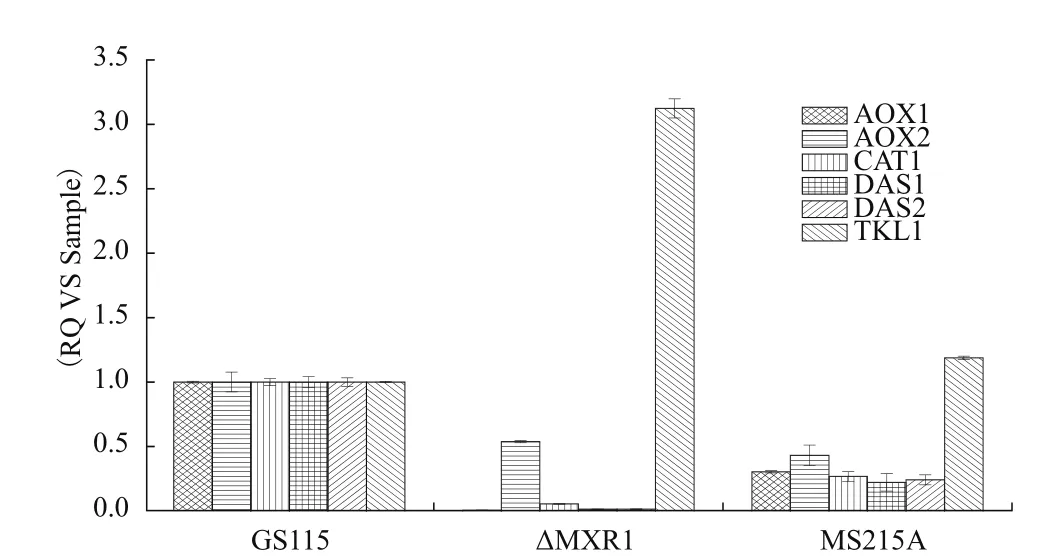
图4 Mxr1磷酸化修饰对甲醇代谢相关基因表达的影响
猜你喜欢
环境工程技术学报(2022年3期)2022-06-05
中国土壤与肥料(2021年5期)2021-12-02
昆钢科技(2021年6期)2021-03-09
科学(2020年2期)2020-08-24
天津科技(2020年4期)2020-05-09
山东化工(2019年18期)2019-10-16
科技资讯(2018年16期)2018-10-26
中国组织化学与细胞化学杂志(2017年1期)2017-06-15
科技资讯(2017年12期)2017-06-09
天然产物研究与开发(2016年6期)2016-06-05
