
莠去津降解菌泛基因组测序及比较基因组分析
2019-07-29 02:50王娅丽朱姗姗杨峰山马玉堃付海燕刘春光
生物技术通报 2019年7期
王娅丽 朱姗姗 杨峰山 马玉堃 付海燕 刘春光
(1. 黑龙江大学农业微生物技术教育部工程研究中心,哈尔滨 150500;2. 黑龙江大学生命科学学院 黑龙江省寒地生态修复与资源利用重点实验室,哈尔滨 150080;3. 黑龙江大学生命科学学院 黑龙江省普通高校微生物重点实验室,哈尔滨 150080)
莠去津又名阿特拉津,是一种选择性内吸传导型苗前苗后除草剂[1],主要通过植物根部吸收并向上传导,抑制杂草的光合作用使其枯死[2],由于其成本较低,使用方便,价格低廉效果好,已成为世界范围内广泛使用的三嗪类除草剂,在美国的年使用量高达36 000 t[3],主要用于玉米、甘蔗、高粱田和草坪杂草的防除。据报道,2004年莠去津被欧盟正式禁止使用,但美国、印度和中国等农业大国仍在使用[4-6]。莠去津施用后大部分进入土壤,其在环境中结构稳定,半衰期为56-364 d,被微生物代谢分解的过程缓慢,在垦区的一些农场发生莠去津后茬敏感作物减产甚至绝产的药害情况,严重影响了农业种植结构的调整和农业生产的安全[7-10]。因此寻找一种有效解决莠去津残留污染的方法迫在眉睫。生物修复是目前用来消除环境污染物的有效治理方法,已成为当前国内外环境科学研究的热点。国内外科研人员相继筛选出多种降解莠去津的土壤微生物[11],结果发现细菌、真菌、放线菌和藻类中均有能降解莠去津的菌株[12]。
1995年,Mandelbam[13]从长期施用莠去津的土壤中分离到第一株以莠去津为唯一氮源的革兰氏阴性假单胞菌Pseudomonassp. ADP[14],降解基因阐明后,成为莠去津降解微生物的模式菌株。此后,研究人员又发现了一株革兰氏阳性菌Arthrobacter aurescensTC1[15],并克隆了其降解基因,然而这两株菌的降解基因并不完全一致。近期有一些莠去津降解菌的相关报道,无论是利用普通PCR扩增还是高通量测序均发现不同种属细菌的降解相关基因型也并不完全一致,这可能与不同种属菌株的降解特性有关[16-18]。
本课题组从黑龙江省长期施用莠去津的玉米田土壤中成功分离出以该除草剂为唯一碳源生长的高效降解菌株Microbacteriumsp. HBT4,该菌108 h对于100 mg/L的莠去津的降解率能够达到99%以上,为黑土农田莠去津污染土壤的修复研究工作提供了良好微生物资源。据报道,Microbacterium属的菌株多见于转化蔗糖、合成低聚果糖、产絮凝剂、产纤维素酶、降解多环芳烃、赤霉烯酮、重金属等研究[19-23],然而作为莠去津的降解菌株却少有报道,且未见该菌比较基因组学分析相关报道,可能与文献报道的基因型不同,也可能是由于适应环境而产生若干突变,存在基因水平转移现象,有待进一步研究。为更加深入了解该菌株与已报道的两株模式菌株基因组的异同,本研究对降解菌HBT4进行泛基因组测序,与GenBank中已登录的两株模式菌株进行比较基因组分析,旨在找到3株菌株参与莠去津降解的代谢途径,为明确菌株HBT4降解莠去津的相关分子机理奠定基础。
1 材料与方法
1.1 材料
供试菌株:菌株HBT4是从黑龙江省长期施用莠去津的玉米田土壤中分离纯化出的降解菌。菌株Arthrobacter aurescensTC1和Pseudomonassp. ADP的基因组数据来源于NCBI数据库(https://www.ncbi.nlm.nih.gov/)。
1.2 方法
试验流程按照Illumina公司提供的标准规程执行,在Illumina Hiseq 4000测序平台对菌株HBT4进行测序,包括DNA小文库制备和测序试验。首先提取样品DNA,其次使用超声波随机打断,获得需要的插入片段,然后在片段的粘性末端添加引物构建文库,将构建好的测序文库在测序芯片上进行桥式PCR 后进行双端测序,最后将产生的数据解读成核酸序列用于下一步信息分析。
对测序产生的原始reads进行评估,再通过过滤低质量、纠错等方法得到高质量的reads,然后使用Velvet软件[24]对菌株HBT4的测序数据进行组装,通过软件Prodigal[25]对组装后的基因组进行编码基因预测,使用Repeat Masker[26]软件对细菌基因组进行重复序列预测,使用软件tRNAscan-SE[27]预测基因组中的tRNA,使用软件Infernal 1.1[28]基于Rfam[29]数据库预测基因组中的rRNA,通过编码基因、重复序列和非编码RNA预测对基因组组分进行分析,使用通用数据库和专用数据库对基因功能进行注释,通过SNP、Small InDel SV检测水平转移基因获得基因间变异,通过核心基因组、物种特有基因和共线性分析与系统进化树构建进行比较基因组学分析。
通用数据库注释方法如下:利用预测得到的基 因 序 列 与 COG[30]、KEGG[31]、Swiss-Prot[32]、TrEMBL[32]、Nr[33]等功能数据库做 BLAST[34]比对,得到基因功能注释结果。基于nr数据库比对结果,应用软件 Blast2GO[35]进行 GO[36]数据库的功能注释。利用软件HMMER基于Pfam[37]数据库进行Pfam功能注释。另外根据注释结果进行进一步的COG类别分析、KEGG代谢通路分析和GO功能分类分析。专用数据库注释方法如下:利用预测得到的基因的蛋白序列与转运蛋白分类数据库(TCDB)[38]、病原体-宿主互作因子数据库(PHI)[39]、抗生素抗性基因数据库(ARDB)[40]、毒力因子数据库(VFDB)[41]等功能数据库做 BLAST[34]比对,得到相应的注释结果。另外利用软件HMMER基于碳水化合物相关酶数据库(CAZyme)[42]进行碳水化合物酶类基因的功能注释。
应用MUMmer[43]软件包将参考菌株与测序菌株基因组进行基因组比对,找到SNP和Small InDel;利用MUMmer软件包将参考菌株与测序菌株基因组进行基因组比对,根据得到的同源共线性区域的信息识别出每一个菌株相比于参考基因组发生结构变异的区域;利用两种软件Alien Hunter[44]和HGT-Finder[45]分别对菌株Arthrobacter aurescensTC1和菌株Pseudomonassp. ADP进行水平转移基因预测;使用软件Mugsy[46]对将所有测序菌株的基因组序列和参考基因组的基因组序列进行比对,从比对结果中找出各菌株共有的序列为核心基因组序列,剩余序列为非必需基因组序列;使用OrthoMCL[47]对各个菌株预测得到的蛋白序列以及参考基因组的蛋白序列进行基因家族分析,寻找每个菌株共有的和特有的基因家族;使用测序物种及参考物种的单拷贝蛋白序列,利用phyML[48]软件构建进化树,研究物种间的进化关系。
2 结果
2.1 测序结果统计
对菌株构建270 bp文库,结果得到测序数据量为0.92 Gb,测序深度为260X,测序质量值在20以上的碱基比例为96.98%,测序质量值在30以上的碱基比例为92.24%。
2.2 基因组组装结果
组装得到的基因组为3.53 Mb,Scaffold片段数量为10,Scaffold N50的长度为0.67 Mb,Contig 片段数量为17,Contig N50的长度为0.62 Mb,GC含量为69.98%。
2.3 基因组组分分析
菌株HBT4预测到的基因个数为3 397,所有基因的长度和为3.26 Mb,平均每个基因的长度为959 bp。菌株HBT4预测出的重复序列总长度为46 967 bp,基因组中重复序列含量为1.33%。非编码RNA即不编码蛋白质的RNA,针对非编码RNA的结构特点,采用不同的策略预测不同的非编码RNA。预测到的tRNA数量为48,预测到的种类数量为43;预测到的rRNA数量为6,预测到的种类数量为3;预测除tRNA和rRNA之外的其它ncRNA,预测到的rRNA数量为9,预测到的种类数量为8。
2.4 基因功能注释
2.4.1 通用数据库注释 通用数据库功能注释统计信息如表1所示。
2.4.2 专用数据库注释 注释结果的统计如表2所示。
2.5 菌株间变异差异分析
结果显示,菌株Arthrobacter aurescensTC1的SNP碱基个数为3 114,Small InDel碱基个数为132,菌株Pseudomonassp. ADP的SNP碱基个数为624,Small InDel碱基个数为48;菌株Arthrobacter aurescensTC1和菌株Pseudomonassp. ADP的Deletion及Insertion的结构变异数量均为0;菌株Arthrobacter aurescensTC1两种软件均预测是水平转移基因的基因数量为77个,两种软件预测到的水平转移基因的并集为1 509,菌株Pseudomonassp. ADP两种软件均预测是水平转移基因的基因数量为24,两种软件预测到的水平转移基因的并集为1 301。
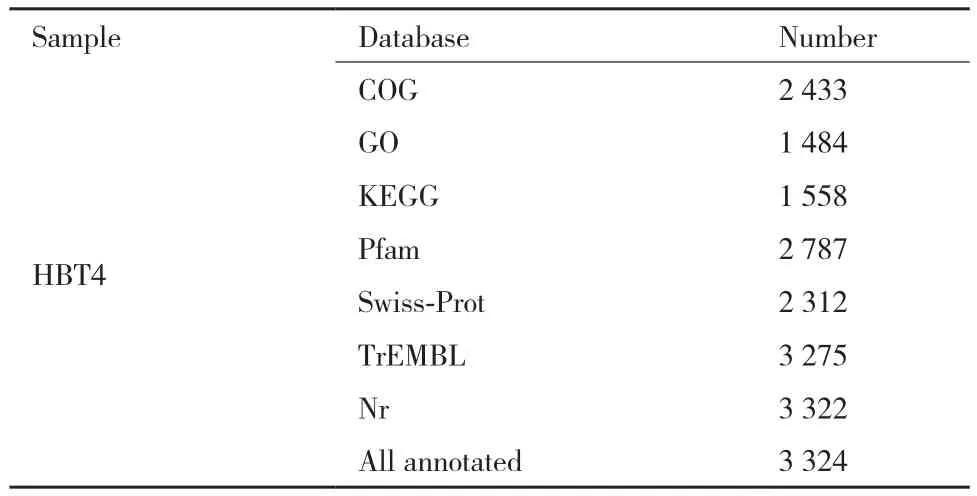
表1 基因功能注释统计信息

表2 基因功能注释统计信息
2.6 莠去津降解菌间比较基因组分析结果
2.6.1 核心基因组与非必需基因组 比对结果统计显示核心基因组序列占总基因组大小的比例为0.02%,基因组序列长度为2 769 bp;非必需基因组序列占总基因组大小的比例为99.98%,基因组序列长度为12.45 Mb。任意两个菌株共有而其他基因组没有的基因组序列占总基因组大小的比例为0.75%,基因组序列长度为93 264 bp;单个菌株特有的基因组序列占总基因组大小的比例为99.25%,基因组序列长度为12.35 Mb。菌株HBT4占总基因组大小的比例为0;Arthrobacter aurescensTC1占总基因组大小的比例为58.44%,基因组序列长度为7.22Mb;Pseudomonassp. ADP占总基因组大小的比例为41.56%,基因组序列长度为5.13 Mb。
核心基因组与非必需基因组比例分布图如图1所示(图中Germs代表菌株HBT4)。多个菌株的泛基因组中各组分的比例分布图,左侧饼图展现的是泛基因组中核心基因组占泛基因组的比例以及非必需基因组占泛基因组的比例。中间饼图展现的是非必需基因组中,不同个数菌株共有(但不是所有菌株共有)的非必需基因组以及单个菌株特有的非必需基因组各占非必需基因组的比例。右侧矩形展现的是各个菌株分别特有的基因组占物种特有基因组的比例。
2.6.2 核心基因家族和物种特有基因 各菌株共有的基因家族即为核心基因家族,核心基因家族和非必需基因家族的统计结果如图2所示。Microbacteriumsp. HBT4与Arthrobacter aurescensTC1有1891个相同基因家族,Pseudomonassp. ADP与Microbacteriumsp. HBT4有1115个相同基因家族,Arthrobacter aurescensTC1与Pseudomonassp. ADP有1341个相同基因家族,它们之间共同的基因家族有986个。
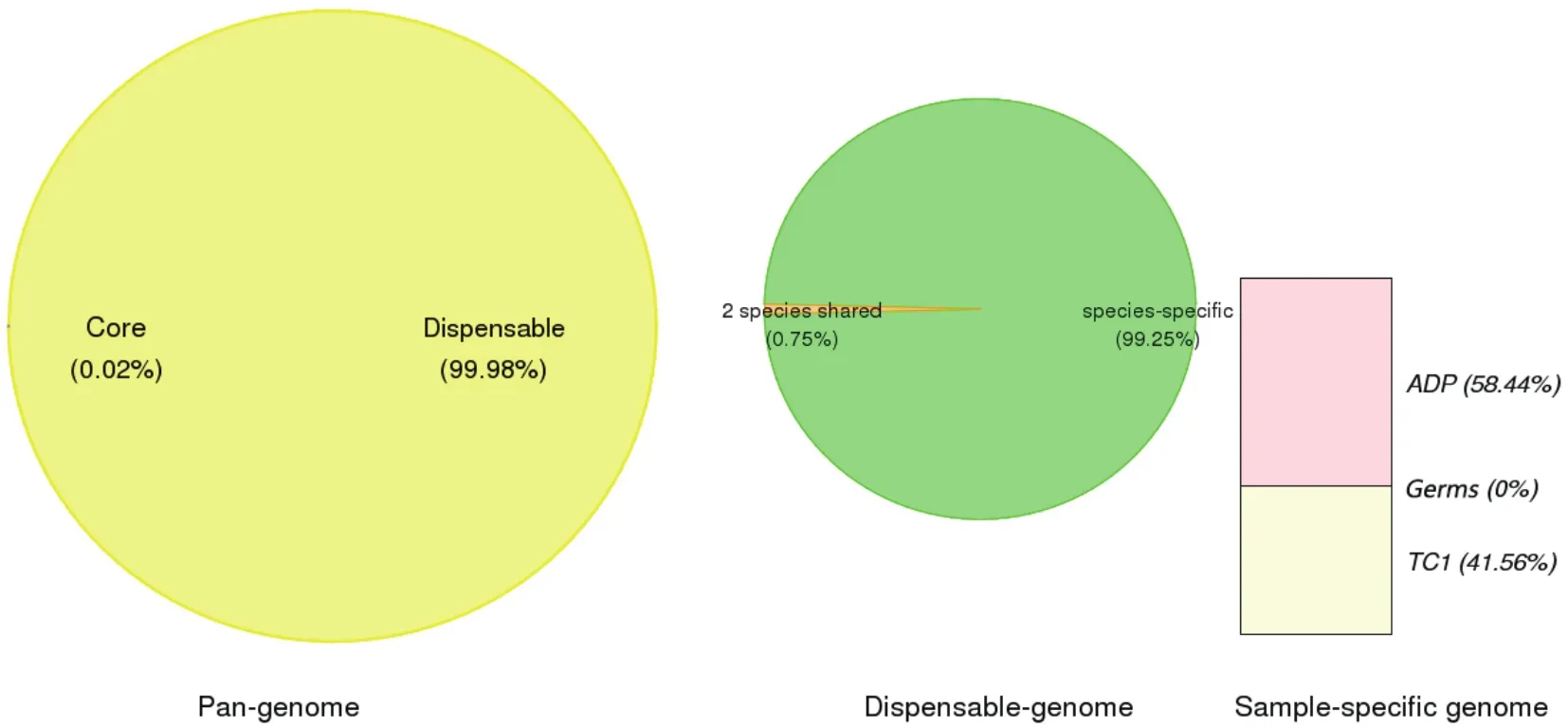
图1 基因组与非必需基因组比例分布图
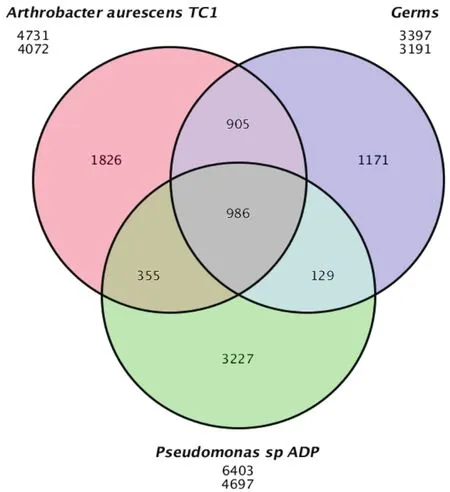
图2 基因家族韦恩图
物种特有基因——泛基因基因家族随物种的添加变化情况为菌株HBT4随着物种由上至下的添加核心基因组中基因家族的数量为1 891,随着物种由上至下的添加泛基因组中基因家族的数量为5 372;Arthrobacter aurescensTC1随着物种由上至下的添加核心基因组中基因家族的数量为4 072,随着物种由上至下的添加泛基因组中基因家族的数量为4 072;Pseudomonassp. ADP随着物种由上至下的添加核心基因组中基因家族的数量为986;随着物种由上至下的添加泛基因组中基因家族的数量为8 599。将数据做成直方图方便比较二者之间的关系如图3所示。该图是由上面的统计表格绘制而成的,沿着纵轴逐渐添加菌株,核心基因家族的数量逐渐减少(砖红色矩形),泛基因家族的数量逐渐增加(蓝色矩形)。
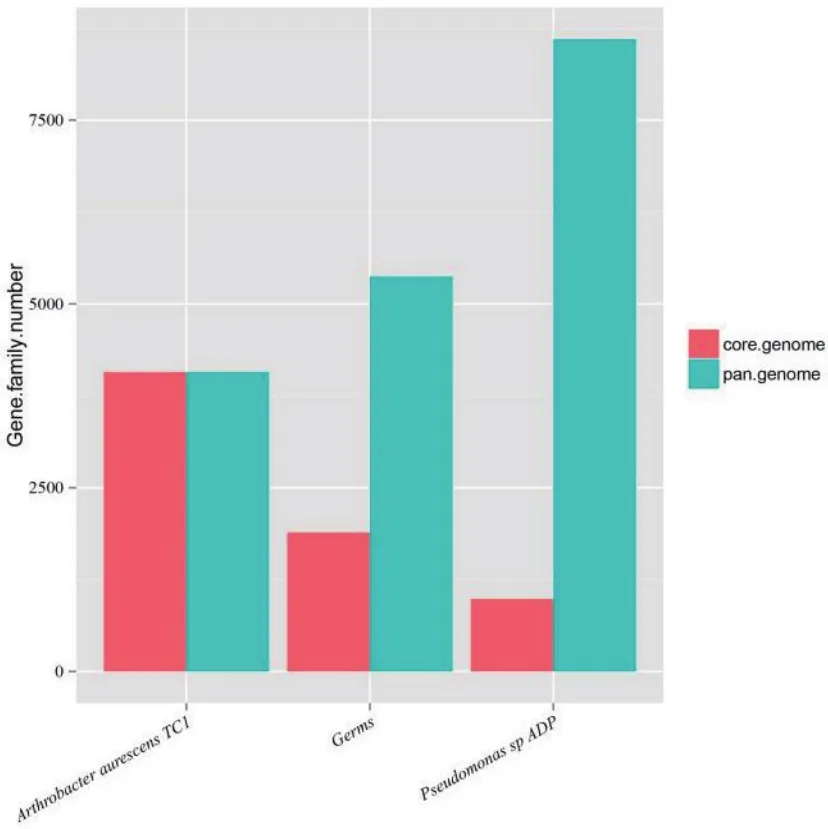
图3 泛基因家族随物种的添加量变化
对于每个菌株的特有基因(物种特有的基因家族的基因)进行GO数据库注释分析,结果如图4所示。细胞膜上的特有基因在细胞组分GO分类中所占比例最大,具有催化功能的特有基因在分子功能GO分类中所占比例最大,代谢途径中的特有基因在生物进程GO分类中所占比例最大。
经过泛基因组测序和比较基因组分析,发现共有基因和特有基因主要集中在三羧酸循环KEGG代谢通路中,如图5所示。三羧酸循环KEGG代谢通路中的共有基因和特有基因功能注释如表3所示。
2.6.3 系统发育树构建 系统进化关系结果如图6所示。能够看出Microbacteriumsp. HBT4与Arthrobacter aurescensTC1进化程度相近,而Pseudomonassp. ADP进化程度较高。
2.6.4 基因共线性分析Microbacteriumsp .HBT4,Arthrobacter aurescensTC1与Pseudomonassp. ADP共线性结果如图7所示。图中左边是相应的菌株名,右边的长矩形框组成的每一行表示一个菌的基因组,每个矩形框表示一个scaffold,不同基因组之间彩色的连线表示上下两个基因组之间同源基因在各个不同细菌基因序列上的不同位置信息得到核酸水平上的共线性关系。从图中可以看出Microbacteriumsp.HBT4与Arthrobacter aurescensTC1基因共线性情况明显高于Pseudomonassp. ADP与Microbacteriumsp.HBT4,所以Microbacteriumsp. HBT4与Arthrobacter aurescensTC1亲缘关系更近一些。
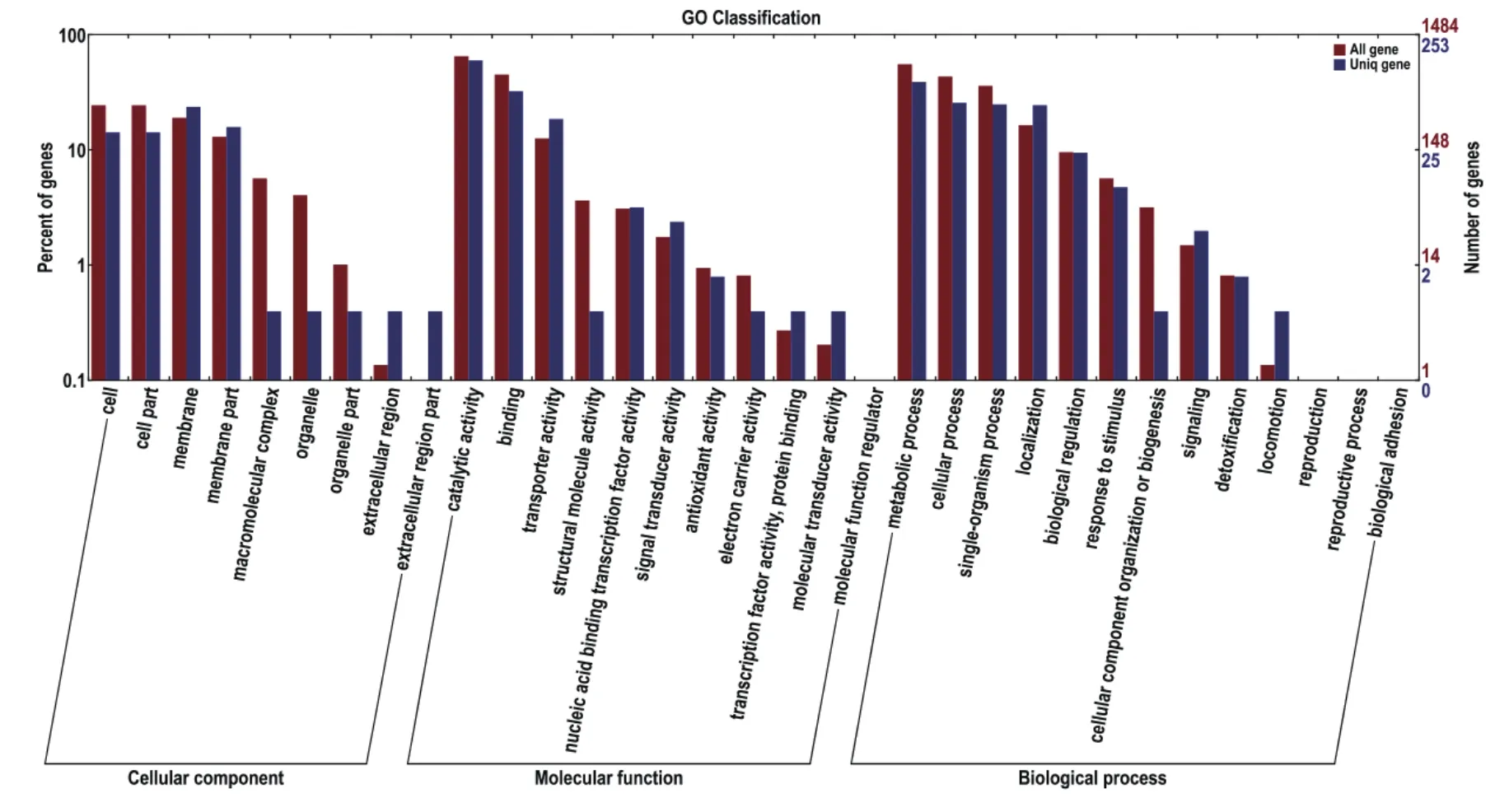
图4 特有性基因的GO二级节点注释分类
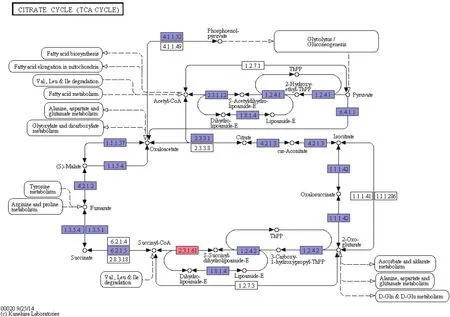
图5 共有和特有基因的KEGG代谢通路富集图
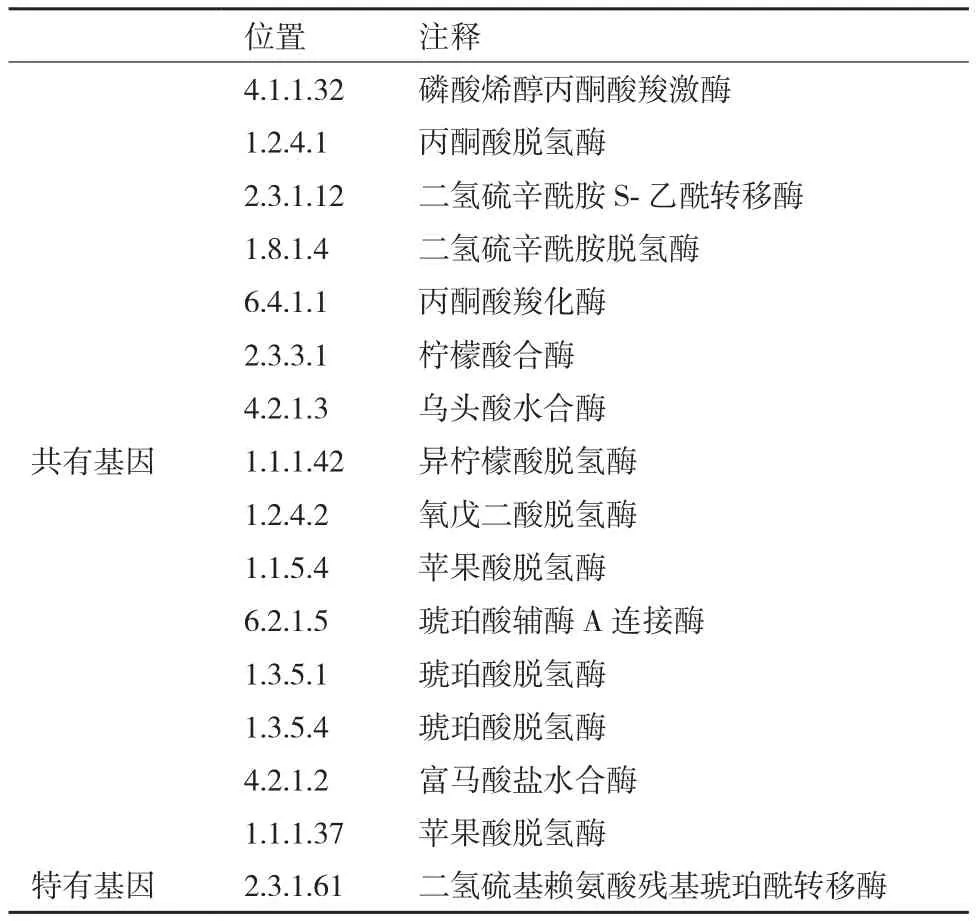
表3 三羧酸循环KEGG代谢通路中的共有基因和特有基因功能注释
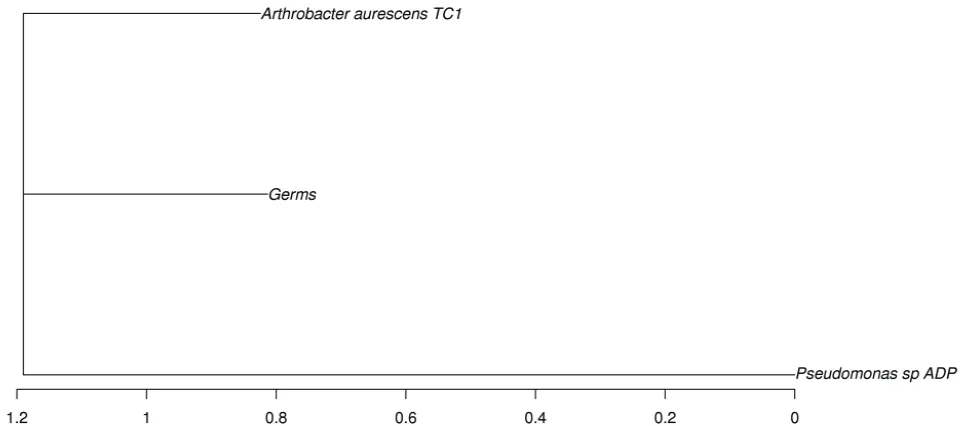
图6 各菌株与参考菌株的系统进化树
3 讨论
据报道细菌对多环芳烃的降解途径最终通过进入三羧酸循环得以完全降解,推测菌株的降解特性与三羧酸循环具有相关性。史延华等[49]初步推测菌株Pseudomonas stutzeriYC-YH1对萘的降解是通过水杨酸途径,萘首先被其代谢为1,2-二羟基萘,而后被转化为水杨酸和邻苯二酚,最后进入三羧酸循环被彻底降解。张丹等[50]综述了恶臭假单胞菌、分枝杆菌属、芽胞杆菌属、微球菌属及粪产碱菌等细菌对菲的降解是通过水杨酸途径或者邻苯二甲酸途径降解,经过一系列反应进一步氧化开环后,最终进入三羧酸循环彻底降解为二氧化碳和水。另外,高亚平等[51]研究表明,莠去津的长期作用改变了鳗草组织氮含量和碳氮比,降低了糖和三羧酸循环中间产物的水平,这说明长期的莠去津暴露会降低能量供应并改变碳氮代谢。
本研究中3个菌株的基因组经比较获得的共有和特有基因KEGG代谢通路,在由丙酮酸生成乙酰辅酶A时,菌株HBT4、ADP和TC1首先在丙酮酸脱氢酶作用下生成2 -羟基乙基ThPP,然后在丙酮酸脱氢酶作用下生成乙酰二氢丙醇酰胺,最后在二氢硫辛酰胺S-乙酰转移酶作用下生成乙酰辅酶A,而不是由丙酮酸在丙酮酸脱氢酶(1.2.7.1)作用下直接脱氢生成乙酰辅酶A,这与经典三羧酸循环稍有差异。在第一次脱氢由异柠檬酸生成α-酮戊二酸时,3株菌都是先在异柠檬酸脱氢酶作用下转化成草酰琥珀酸盐,再经过脱氢生成α-酮戊二酸,而不是直接由异柠檬酸在异柠檬酸脱氢酶(1.1.1.41/1.1.1.286)作用下生成α-酮戊二酸。第2次脱氢时,3株菌都是先在氧戊二酸脱氢酶作用下生成羧基羟丙基ThPP,然后脱氢生成琥珀酰二氢硫辛酰胺,最后在二氢硫基赖氨酸残基琥珀酰转移酶作用下生成琥珀酰辅酶A,而不是α-酮戊二酸在α-酮戊二酸脱氢酶(1.2.7.3)作用下直接生成琥珀酰辅酶A,其中特有基因(2.3.1.61)就位于α-酮戊二酸与琥珀酰辅酶A的代谢通路之间。

图7 各菌株与参考菌株的共线性图
另外,由KEGG通路图可知,α-酮戊二酸与抗坏血酸盐和醛酸盐代谢、丙氨酸、天冬氨酸和谷氨酸盐代谢、D-谷氨酰胺和D-谷氨酸代谢之间存在双向的间接影响;缬氨酸、亮氨酸和异亮氨酸的降解对琥珀酰辅酶A代谢产生间接影响;酪氨酸代谢、精氨酸和脯氨酸代谢对延胡索酸产生间接影响;丙氨酸、天冬氨酸和谷氨酸代谢、乙醛酸盐和二羧酸盐代谢与草酰乙酸之间存在双向的间接影响。缬氨酸、亮氨酸和异亮氨酸的降解还有脂肪酸代谢对乙酰辅酶A有间接影响;乙酰辅酶A对脂肪酸合成和线粒体中脂肪酸延长有间接影响;草酰乙酸在磷酸烯醇丙酮酸羧激酶作用下生成的磷酸烯醇丙酮酸盐对糖酵解/糖异生有间接影响,糖酵解/糖异生对丙酮酸代谢也存在间接影响。
三株菌共有基因所在的代谢途径有可能是其参与莠去津降解的原因,而三羧酸循环中特有基因编码的二氢硫基赖氨酸残基琥珀酰转移酶可能是Microbacteriumsp. HBT4与Pseudomonassp. ADP和Arthrobacter aurescensTC1的不同之处。三羧酸循环的中间产物,从理论上讲可以循环不消耗,但是由于循环中的某些组成成分还可参与合成其他物质,而其他物质也可不断通过多种途径而生成中间产物,所以说三羧酸循环组成成分处于不断更新之中。
4 结论
本研究通过泛基因组测序和比较基因组分析,得到该菌株基因组大小约为3.53 Mb,预测到菌株HBT4编码基因3 397个、重复序列含量为1.33%、非编码RNA 63个,通用数据库基因功能注释共3 324个,专用数据库基因功能注释共1 149个,通过菌株间差异变异分析发现SNP、Small InDel和水平转移基因,未发现结构变异基因,获得该菌株特有基因中GO注释到的基因在细胞组分、分子功能和生物学进程中的数量和比例,从KEGG 代谢通路富集图中发现特有基因编码的二氢硫基赖氨酸残基琥珀酰转移酶位于三羧酸循环中α-酮戊二酸和琥珀酰辅酶A的代谢通路之间。获得3个菌株核心基因组与非必需基因组比例分布、系统进化树和共线性关系,发现三者之间共有基因家族986个、菌株HBT4特有基因家族1 171个。该菌株与两株模式菌株相比,其基因家族之间既有相同之处,又有较大差异。
猜你喜欢
今日农业(2021年11期)2021-08-13
建材发展导向(2021年7期)2021-07-16
建材发展导向(2021年24期)2021-02-12
中国生殖健康(2020年4期)2020-12-09
中西医结合肝病杂志(2020年2期)2020-10-27
小哥白尼(军事科学)(2019年9期)2019-12-21
电影(2019年3期)2019-04-04
中成药(2018年7期)2018-08-04
少儿科学周刊·少年版(2017年3期)2017-06-29
中国塑料(2015年2期)2015-10-14
