
甲基膦酸哪酯分子印迹聚合物的合成与识别特性研究
2019-07-27 07:07李盛菘郑永超钟近艺赵冲林
分析测试学报 2019年7期
李盛菘,郑永超,2,钟近艺,2,赵冲林,2*
(1.中国人民解放军军事科学院 防化研究院,北京 102205;2.国民核生化灾害防护国家重点实验室,北京 102205)
对事件现场采集的样品进行准确分析鉴定是可为验证是否存在化学毒剂使用提供最有力证据,但事故现场残留的样品往往会随着时间和环境影响逐步分解和消失,因此对现场残存的超痕量样品及其降解产物的富集和提纯是成功完成分析鉴定任务的关键。目前使用的萃取和浓缩前处理手段,存在背景干扰大、富集倍数低等问题,给后续仪器分析造成困难,尤其是在有大量化合物干扰、样品浓度极低的情况下,使用常规取样分析方法可能无法检出。分子印迹技术是一种高选择性识别技术,已被广泛应用于异构体和对映体的分离提纯[1-3]、特定分子的传感识别[4-6]、痕量组分的分离富集[7-10]等诸多领域。甲基膦酸哪酯(O-Pinacolyl methylphosphonic acid,PMPA)是化学毒剂梭曼的降解产物,也是合成毒剂的前体,属于公约附表2B化学品[11]。本文通过研究甲基膦酸哪酯的特征官能团,从微观层面揭示了甲基膦酸哪酯分子与功能单体分子的作用机理,通过预组装体系设计,合成出对甲基膦酸哪酯具有特征识别作用的分子印迹聚合物,将该分子印迹聚合物制备成固相萃取小柱,成功应用于复杂样品中超痕量甲基膦酸哪酯的分离富集和分析。
1 实验部分
1.1 试剂及仪器
N,O-双(三甲基硅基)三氟乙酰胺(BSTFA)硅烷化试剂(纯度≥98%,美国Acros公司);甲基丙烯酸(MAA)、丙烯酰胺(AM)、二氯甲烷、甲醇、乙腈、氘代乙腈、四氯化碳(纯度均大于98%,美国Aldrich);偶氮二异丁腈(AIBN)、三羟甲基丙烷三甲基丙烯酸酯(EGDMA)、丙酮、冰醋酸(分析纯,北京化学试剂公司);甲基膦酸哪酯(纯度≥95%,军事科学院防化研究院提供)。
GC 8000气相色谱仪(配FPD-P检测器,美国Thermo Fisher公司);5975C型GC-MS联用仪(美国Agilent公司);VARIAN NMR SYSTEM 600超导核磁共振波谱仪(美国Varian公司);Spectrum GX FTIR System傅立叶变换红外光谱仪(美国Perkin Elmer公司);JSM-6460电子显微镜(日本电子公司);501A型恒温水浴(上海浦东荣丰科学仪器有限公司);真空泵和旋转蒸发仪(德国Heidolph公司);索氏萃取装置(北京欣维尔公司)。
1.2 甲基膦酸哪酯预组装分子印迹体系设计与合成
1.2.1 核磁共振波谱研究甲基膦酸哪酯与功能单体之间的相互作用以氘代乙腈为溶剂,配制50 mg/mL的甲基膦酸哪酯溶液,室温下测定其31P{1H} NMR谱和1H NMR谱。与功能单体甲基丙烯酸(或丙烯酰胺)复配后,测定相应的氢谱和磷谱。比较功能单体加入前后化学位移的变化。逐渐增加核磁管中反应液的温度(从室温25 ℃直至聚合反应时的温度60 ℃),研究温度变化对分子间作用力的影响。
1.2.2 红外光谱研究甲基膦酸哪酯与功能单体之间的相互作用以四氯化碳为溶剂,配制5%(质量分数)的甲基膦酸哪酯溶液,采用液膜制样法,测定溶液的红外光谱图。与功能单体甲基丙烯酸(或丙烯酰胺)复配后,再次测定溶液的红外光谱图。比较功能单体加入前后的谱图变化。
1.2.3 甲基膦酸哪酯预组装分子印迹聚合物的合成在30 mL乙腈中加入10 mmol 甲基膦酸哪酯,再加入40 mmol功能单体,超声10 min溶解,随后于5 ℃下放置12 h。加入交联剂EGDMA 200 mmol和1%引发剂AIBN。超声20 min后将该混合溶剂倒入50 mL自制反应瓶中,充入氮气5 min排氧,抽真空,密封。将反应瓶置于60 ℃恒温水中聚合反应24 h,得到白色的棒状聚合物。取出聚合物研磨,过80目筛,然后用丙酮反复沉降,除去过细粒子,再用甲醇-乙酸(9∶1,体积比)索氏提取24 h,最后用乙腈索氏提取12 h。将提取液浓缩20倍后在GC-MS选择离子流模式下检测模板分子的去除程度。取出分子印迹聚合物,用旋转蒸发仪于40 ℃真空干燥30 min,得到干燥的分子印迹聚合物颗粒。
1.3 甲基膦酸哪酯分子印迹聚合物表征
1.3.1 表观特性分别将未去除模板分子的聚合物和去除模板分子的聚合物用KBr压片,测定红外区4 000~400 cm-1下的吸收谱带,研究印迹分子聚合物的官能团特点。
将未去除模板分子的聚合物和去除模板分子的聚合物进行喷金处理,用电子显微镜观测聚合物的粒径大小及形貌。
1.3.2 吸附特性准确称取一定量的分子印迹聚合物于4 mL螺口样品小瓶中,加入一定体积的甲基膦酸哪酯溶液,振荡,放置10 min。分析吸附后清液中剩余的甲基膦酸哪酯的浓度,根据吸附前后模板分子浓度的变化,计算出平均结合量Q,绘制吸附等温线。
1.3.3 识别特性以甲基膦酸哪酯的同系物或结构相近的化合物作为交叉选择性的目标化合物,并配制成一定浓度的样品溶液。准确称取100 mg相应的分子印迹聚合物于4 mL螺口样品小瓶中。加入一定体积的交叉选择性目标化合物标准溶液,振荡,放置10 min。取清液分析,根据吸附前后各种化合物的浓度变化计算吸附量,并用静态分配系数和识别因子评价分子印迹聚合物的交叉选择性。
1.4 分子印迹聚合物用于复杂样品中痕量甲基膦酸哪酯的萃取分离
1.4.1 固相萃取小柱的制作在一支体积为30 mL的聚丙烯固相萃取小柱(63 mm×8.9 mm)中放入一孔径为10 μm的聚乙烯筛板,准确称取300 mg的分子印迹聚合物填入其中,再放入一个相同尺寸的聚乙烯筛板固定,得到填充均匀、柱压降适中的分子印迹聚合物固相萃取小柱,备用。普通硅胶萃取柱制备方法相同,只是填料为普通硅胶。
1.4.2 复杂样品中痕量甲基膦酸哪酯的萃取分离将1 μg/mL甲基膦酸哪酯的乙腈溶液和相同目标物浓度下加入1 mg/mL柴油干扰的乙腈溶液进行硅烷化衍生,待GC-MS分析。取上述被干扰的样品溶液2 mL,以“1.4.1”中制作的分子印迹固相萃取小柱进行萃取分离,具体步骤为:将被干扰的乙腈溶液上样后,采用5 mL二氯甲烷作淋洗剂,最后用2 mL甲醇洗脱目标物,将收集的洗脱液进行衍生与分析操作。将分子印迹固相萃取小柱替换为普通硅胶填料固相萃取小柱进行同样的上样-淋洗-洗脱-衍生-分析步骤。对比4个分析结果,考察分子印迹固相萃取对复杂样品中痕量甲基膦酸哪酯的萃取分离能力。
2 结果与讨论
2.1 分子间作用力研究
2.1.1 从化学结构推导分子间作用力模板分子和功能单体在聚合前能否形成稳定的复合物,是获得高选择性和高亲和性分子印迹聚合物的关键。甲基膦酸哪酯分子中,氧原子核外层有6个电子,两个2s轨道上有4个电子,另外2个孤电子占据p轨道。磷原子外层有5个电子,一个3s轨道上有2个电子,3个p轨道上有3个电子,另外有5个空的d轨道,与氧形成双键时,磷进行sp3杂化,呈四面体构型,可形成4个δ键,其中1个与氧上的2px形成1个δ键,另外3个和烷基及烷氧基形成3个δ键。磷具有利用3d空轨道成键的能力,与氧的2pz轨道形成d-p的π键,这样在磷和氧之间形成1个δ和1个π键的双键,但是磷的电负性(2.1)远小于氧的电负性(3.5),这就造成电子云向氧一侧靠近[12],因此氧上带有的部分负电荷,可以和亲电试剂形成氢键。同时,甲基膦酸哪酯分子中还含有1个羟基,整体显示酸性(PKa=4.7),羟基上的氢也可以和电负性强的化合物形成氢键或者离子键。图1为甲基膦酸哪酯与甲基丙烯酸形成氢键、与丙烯酰胺形成离子键的示意图。

图1 甲基膦酸哪酯与功能单体之间的成键示意图Fig.1 Bonding diagram between O-pinacolyl methylphosphonic acid and functional monomer moleculesleft:methylacrylic acid,right:acrylamide(左为甲基丙烯酸,右为丙烯酰胺)
2.1.2 从化学位移研究分子间作用力图2左图为甲基膦酸哪酯在氘代乙腈(左A),以及在氘代乙腈中跟丙烯酰胺(AM,左B)、甲基丙烯酸(MAA,左C)混合时的31P{1H}NMR谱。结果显示,在氘代乙腈溶液中磷的化学位移是33.973,加入功能单体AM后,因AM中的氮原子提供了电子,形成离子键,增加了磷原子周围的电子云密度,起到屏蔽作用,使磷的化学位移向高场移动至32.802;而加入功能单体MAA后,因氢键的形成,磷核周围的电子云密度降低,起到去屏蔽作用,化学位移向低场移动至35.721。图2右图A为甲基膦酸哪酯的1H NMR谱,测定结果和31P{1H}NMR一致;在功能单体AM加入后,甲基膦酸哪酯上的活泼氢,因与AM的氮作用而消失(右图B);而加入MAA后形成氢键,平均化学位移向低场移动了近6个化学位移单位(右图C)。
另外,体系温度升高到60 ℃,化学位移基本不变。即在加热增加分子运动能量时,分子间的这种作用力相对固定。
2.1.3 从振动频率研究分子间作用力红外光谱上,磷氧双键伸缩振动频率依赖于磷上的取代基[13],由π常数表示:
νP=O=930 + 40∑π
(1)
查表得πCH3=2.1,πO-CH2=2.85,πOH=1.9,代入上式得νP=O为1 204。
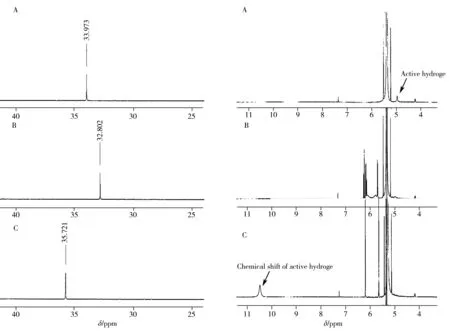
图2 甲基膦酸哪酯的31P{1H}NMR谱(左)和1H NMR谱(右)Fig.2 31P{1H}NMR spectroscopy(left) and 1H NMR spectroscopy(right) of O-pinacolyl methylphosphonic acidA.the deuterated acetonitrile solution of O-pinacolyl methylphosphonic acid(0.33 mol/L);B.the deuterated acetonitrile solution of O-pinacolyl methylphosphonic acid(0.33 mol/L) and acrylamide( AM) functional monomer molecule(1.33 mol/L);C.the deuterated acetonitrile solution of O-pinacolyl methylphosphonic acid(0.33 mol/L) and methylacrylic acid(MAA) functional monomer molecule(1.33 mol/L)(A.甲基膦酸哪酯的氘代乙腈溶液(0.33 mol/L);B.甲基膦酸哪酯在氘代乙腈中(0.33 mol/L)与单体AM(1.33 mol/L)混合;C.甲基膦酸哪酯在氘代乙腈中(0.33 mol/L)与单体MAA(1.33 mol/L)混合)

图3 甲基膦酸哪酯的FT-IR谱Fig.3 FT-IR spectroscopy of O-pinacolyl methylphosphonic acidA.O-pinacolyl methylphosphonic acid in carbon tetrachloride;B.O-pinacolyl methylphosphonic acid in carbon tetrachloride with acrylamide(AM) functional monomer molecule;C.O-pinacolyl methylphosphonic acid in carbon tetrachloride with methylacrylic acid(MAA) functional monomer molecule(A.在四氯化碳中;B.在四氯化碳中与单体AM混合;C.在四氯化碳中与单体MAA混合)
2.2 甲基膦酸哪酯分子印迹聚合物表征
2.2.1 形貌表征按“1.2.3”合成的甲基膦酸哪酯预组装分子印迹聚合物(AM为单体)为白色的棒状聚合物,经研磨过筛,用甲醇-乙酸(9∶1,体积比)索氏提取24 h除去模板分子,得到甲基膦酸哪酯分子印迹聚合物颗粒。
扫描电镜结果显示,所得分子印迹聚合物为粒径几十微米的颗粒(图4A)。将其中一个颗粒局部表面分别放大至2万倍和5万倍,发现其为多孔性材料(图4B、C),这些孔使分子印迹聚合物的比表面积大大增加,从而增加了孔壁表面印迹点位的数量。
2.2.2 红外光谱表征采用傅立叶变换红外光谱仪测定以AM为功能单体合成的未去除模板分子的聚合物和去除模板分子的聚合物(KBr压片法)在4 000~400 cm-1下的吸收谱带,结果见图5。
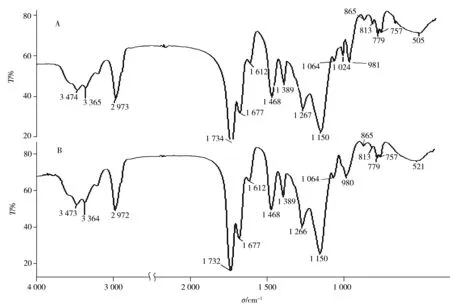
图5 甲基膦酸哪酯分子印迹聚合物的吸收光谱图Fig.5 Absorption spectra of O-pinacolyl methylphosphonic acid-imprinted polymerA.with template molecule;B.without template molecule(A.未去除模板分子;B.去除模板分子);AM as monomer

2.2.3 吸附特性表征实验对功能单体为MAA的甲基膦酸哪酯分子印迹聚合物2P3、功能单体为AM的甲基膦酸哪酯分子印迹聚合物2P5及空白分子印迹聚合物2P4进行了研究。分别配制5.0、10.0、20.0、30.0、50.0、80.0、150、200 μg/mL的甲基膦酸哪酯乙腈溶液。准确称取200 mg 分子印迹聚合物8份于4 mL螺口样品小瓶中,分别加入上述8种不同质量浓度的模板分子溶液2 mL,振荡,放置10 min后取清液1 mL,衍生,加入内标,以GC-MS分析吸附后清液中剩余的甲基膦酸哪酯的浓度,计算出结合量Q,结果见表1。

表1 甲基膦酸哪酯分子印迹聚合物对模板分子的吸附量随浓度的变化Table 1 The dependence of absorption capacity of O-pinacolyl methylphosphonic acid-imprinted polymer on template molecule on concentration
以起始浓度为横坐标,吸附量为纵坐标,作2P3、2P5和2P4的吸附等温线,见图6。从吸附等温线上看,随着模板分子质量浓度逐渐增加,2P4和2P5都有吸附饱和的趋势,而2P3没有明显的饱和现象,出现非特异性吸附,分子印迹聚合物2P5的吸附量比2P4大。另外,甲基膦酸哪酯质量浓度低于80.0 μg/mL时,2P5对模板分子呈现一定的线性结合,而2P4的线性不明显。说明在2P5合成过程中,甲基膦酸哪酯与AM形成了复合物,当洗脱甲基膦酸哪酯后在聚合物内部形成了形状与官能团位置均与甲基酸频哪酯相匹配的空穴,甲基膦酸哪酯能与这些空穴发生选择性结合,在空穴被全部结合之前,结合是线性的,空穴全部被甲基膦酸哪酯分子占据后,呈非选择性吸附,直到吸附达到饱和。2P4由于没有这样的空穴,仅呈非选择性结合模式,结合量少。

图6 甲基膦酸哪酯分子印迹聚合物的吸附等温线Fig.6 Absorption isotherms of O-pinacolyl methylphosphonic acid-imprinted polymermonomer molecules are methacrylic acid for 2P3,acrylamide for 2P5 and acrylamide for non-imprinted sol-gel polymer 2P4(2P3功能单体为甲基丙烯酸,2P5功能单体为丙烯酰胺,2P4为空白印迹聚合物)
以选择性结合作用的分子印迹聚合物2P5进行Scatchard模型分析[14]。Scatchard模型表达式为:
Q/Ce=(Qmax-Q)/Kd
(2)
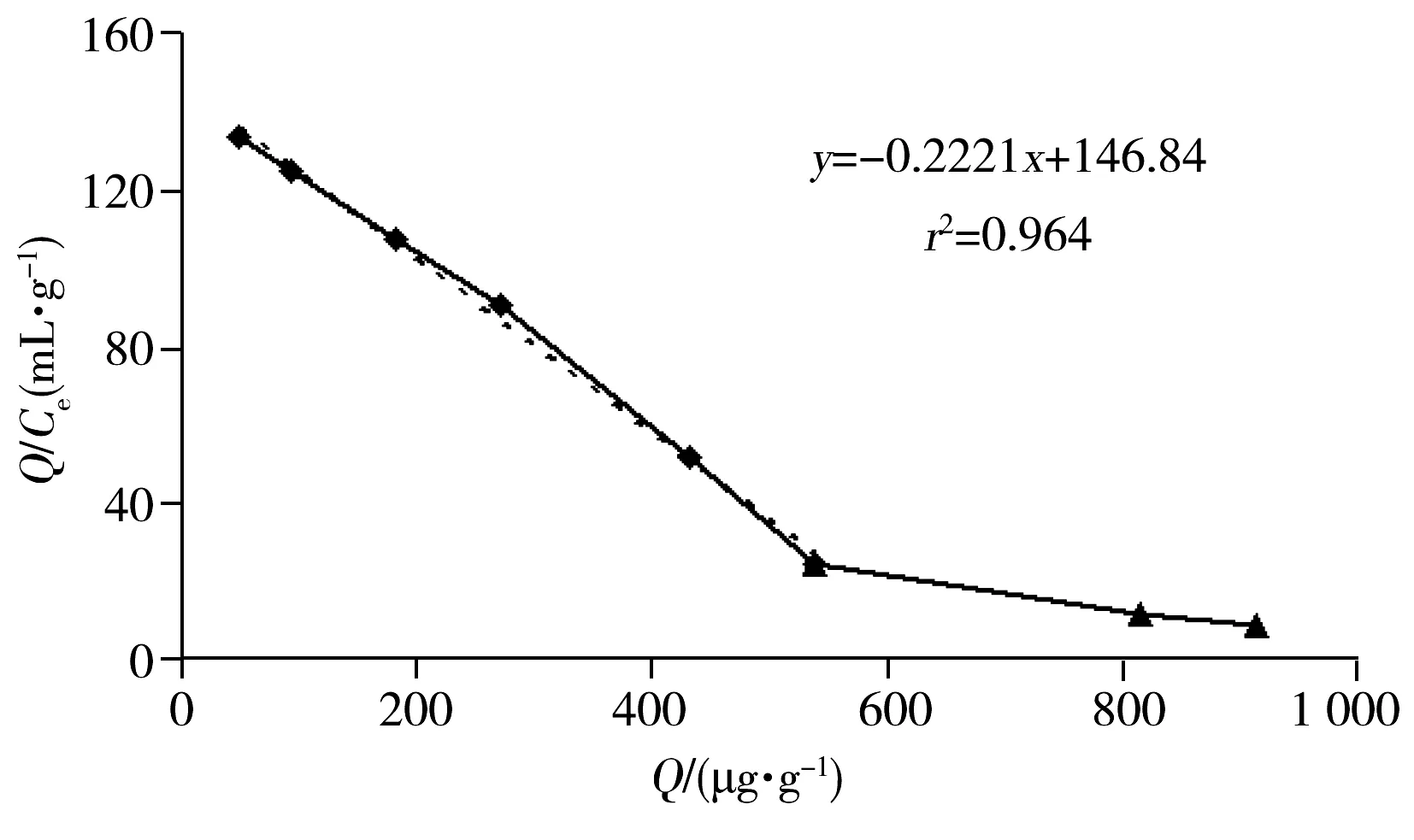
图7 甲基膦酸哪酯分子印迹聚合物的Scatchard分析图Fig.7 Scatchard diagram of O-pinacolyl methylphosphonic acid-imprinted polymer
其中,Q是吸附量,μg/g;Ce是吸附平衡时的模板质量浓度,μg/L;Qmax是结合位点的最大表观结合数,μg/g;Kd是结合位点的平衡常数,μg/L。以Q/Ce为纵坐标,Q为横坐标作图,其Scatchard分析图见图7,在低吸附量范围内(Q<500 μg/g)两者呈明显线性相关。
将表1中的低吸附范围数据代入公式(2),进行线性回归,回归方程为Q/Ce=147-0.222 1Q。即-1/Kd=-0.222 1,计算得解离常数Kd=4.5×10-3μg/L,由Qmax/Kd=147,得最大表观结合量Qmax,AM=661 μg/g。下文均以AM为单体合成的分子印迹聚合物为对象进行研究。
2.2.4 选择特性表征选择性吸附是印迹聚合物最显著的特征之一,分子印迹聚合物洗脱模板分子后留下了与其形状与结合部位相对应的空穴,此空穴可对模板分子进行选择性识别。可用静态吸附分配系数Kp和识别因子α来表征[15]印迹聚合物的选择性。
(3)
式中,Cp表示模板分子在印迹聚合物上的吸附量(μg/g),Cs则表示底物在溶液中的质量浓度(μg/mL)。识别因子α定义为:
(4)
其中Kpi和Kpj分别表示i物质和j物质的静态吸附分配系数。j一般表示模板分子,而i为一般底物,即被用来评价分子印迹聚合物识别特性的其它物质。
表2 甲基膦酸哪酯分子印迹聚合物对8个底物的静态吸附分配系数及识别因子Table 2 Static adsorption distribution coefficient and identification factor of O-pinacolyl methylphosphonic acid-imprinted polymer on eight substrates
2.3 甲基膦酸哪酯分子印迹聚合物用于分离提纯

3 结 论
猜你喜欢
陶瓷研究(2022年3期)2022-08-19
煤化工(2022年3期)2022-07-08
云南画报(2021年10期)2021-11-24
色谱(2021年7期)2021-06-07
小学生优秀作文(高年级)(2018年4期)2018-09-11
中国军转民(2017年7期)2017-12-19
中国资源综合利用(2016年10期)2016-01-22
大连工业大学学报(2015年4期)2015-12-11
中国摄影(2014年12期)2015-01-27
中国卫生(2014年10期)2014-11-12
