
轨道离子阱快速筛查及定量分析保健食品中73种非法添加成分
2019-07-26 00:33王会霞朱晓玲吴婉琴刘国姣
食品与机械 2019年7期
韩 智 龚 蕾 王会霞 朱晓玲 吴婉琴 刘国姣 王 彬
(1.湖北省食品质量安全监督检验研究院,湖北 武汉 430070;2.湖北省食品质量安全检测工程技术研究中心,湖北 武汉 430070)
近年来,中国保健食品产业发展迅速,2018年全国保健食品市场产值达到4 000亿元[1]。一些不法分子为获取高额利润在保健食品中非法添加化学药物,以增强其宣称的功效,可能导致消费者发生轻微中毒,严重的甚至危及生命[2]。据统计[1],2017年7月~2018年5月查处的保健食品违法行为中,非法经营行为占全部案件总数高达45.19%,其中又以非法添加药物为主。
为遏制保健食品中非法添加西药的行为,在2018版《国家食品安全监督抽检实施细则》[3]中,共有67种不同种类的非法添加西药被列入日常监督抽检范围,涉及12种不同检验方法,但每种检验方法的检测成分较少。保健食品非法添加西药常用的检测方法有薄层色谱法[4-6]、酶联免疫法[7-8]、HPLC法[9-11]、GC-MS法[12-13]、HPLC-MS/MS[14-16],HPLC-TOF-MS[17-20]等,这些方法的开发对保健食品中非法添加西药监测提供很好的依据,但存在基质背景干扰问题。
静电场轨道阱(Orbitrap)技术是由俄国科学家[21]根据静电场轨道阱发展的新型质量分析器,具有高分辨率和高质量精度的特点,可实现食品中有毒有害物质或功效成分等物质的高通量筛查,已逐渐应用于保健食品检测中。张晓光等[22]利用Q-Exactive Orbitrap筛查保健食品中23种降压非法添加西药成分,各药物检出限为0.2~0.5 μg/kg;刘芸等[23]利用Q Exative Orbitrap对4类共31种保健食品进行42种非法添加西药进行了筛查及定量测定,检出限为1.0~10.0 ng/mL,检出其中3种添加了非法药物。
目前采用高分辨质谱对保健食品中非法添加物的筛查研究较少,且基本都是某一类或者几类样品单独分析[22-23],有必要建立一个高通量筛查方法,提高检测效率。本试验拟对73种保健食品中非法添加物作为研究对象,旨在建立一种简单且快速的高通量定性筛查和准确定量方法。
1 材料与方法
1.1 材料与试剂
保健食品:市售;
西布曲明、N-单去甲基西布曲明、N-N-双去甲基西布曲明等73种化学药物标准品:纯度≥95%,中国食品药品检定研究院;
乙腈:色谱级,德国Merck公司;
甲酸:色谱级,美国Thermo Fishers公司。
1.2 仪器与设备
高分辨液质联用仪:Orbitrap Fusion型,美国Thermo Fisher公司;
色谱柱:Waters Acquity UPLC HSS T3型,100 mm×2.1 mm,1.8 μm,美国Waters公司;
超纯水器:Milli-Q型,法国密理博公司;
高速离心机:Avanti JXN-30型,美国Beckman Coulter公司;
电子天平:ME204型,梅特勒—托利多仪器有限公司;
数控超声波清洗器:2600T型,上海安谱科学仪器有限公司。
1.3 方法
1.3.1 样品前处理 参考BJS 201710《保健食品中75种非法添加化学药物的检测》[24],准确称取1 g样品,准确加入10 mL甲醇,涡旋混匀,超声提取15 min,4 500 r/min 离心8 min,取上清液过滤膜,供Orbitrap Fusion高分辨液质联用仪测定。
1.3.2 色谱条件优化
(1)色谱柱条件优化:比较了不同粒径及长度的C18色谱柱,包括Waters Acquity HSS T3(2.1 mm×100 mm,1.8 μm),Waters Acquity HSS T3(2.1 mm×50 mm,1.8 μm),Thermo Accucore aQ(2.1 mm×100 mm,2.6 μm),Thermo Hypersill GOLD(2.1 mm×150 mm,3.0 μm),Acquity UPLC BEH C18(2.1 mm×100 mm,1.7 μm),Acquity UPLC BEH C18(2.1 mm×50 mm,1.7 μm),发现Waters Acquity HSS T3(2.1 mm×100 mm,1.8 μm)柱的峰形较好,对强极性物质保留好,峰形好,分离度高,因此本研究选用此色谱柱。
(2)流动相条件优化:比较了不同流动相及洗脱梯度对化合物的分离效果,最终确定流动相:A为乙腈;B为水(含0.1%甲酸)。梯度洗脱程序:0.0~3.0 min,2% A;3.0~12.0 min,2%~95% A;12.0~15.0 min,95% A;15.0~15.1 min,95%~2% A;15.1~20.0 min,2% A。流速0.3 mL/min;柱温35 ℃;进样体积5 μL。
1.3.3 质谱条件优化 本研究采用Full-scan ddMS2采集数据,为避免漏检化合物,将目标母离子精确分子量预先输入到目标离子框中,优先扫描指定m/z,达到设定阈值后触发二级碎裂;利用四级杆隔离将m/z窗口设置为100~1 000,以减少其他杂质离子进入检测器中。经过系列参数筛选后,仪器将选取符合条件且信号最强的母离子进行子离子扫描,使用Orbitrap质量分析器检测子离子,由此得到高分辨数据。为了使分析物的数据质量达到最优,进行了分辨率、碰撞能量等参数的优化。优化后的参数如下:全扫描分辨率R=120 000、二级扫描分辨率R=60 000,自动增益控制目标离子数(AGC)为5×105,质量偏差窗口为5×10-6,高能碰撞裂解(HCD)碎裂能量为(45±15)%,离子源温度325 ℃,离子传输杆温度350 ℃,鞘气265 kPa,辅助气70 kPa。
2 结果分析
2.1 筛查方法建立
按照1.3的方法优化后,73种化合物的保留时间、母离子、碎片离子等信息见表1。在优化的质谱色谱条件下,73种化合物在20 min内都得较好的分离。尽管有一些化合物保留时间较为相同,如西布曲明和去甲基他达那非,保留时间同为8.54 min,但是质荷比有差异,使得很容易区分,见图1。一些化合物为同分异构体,如伪伐地那非和那莫西地那非为同分异构体,质荷比相同,但是保留时间不一致,伪伐地那非保留时间为9.52 min,那莫西地那非为10.57 min,通过保留时间差异,也可进行准确定性分析,见图2。
利用优化好的质谱条件采集标准品数据,采用Xcalibur软件提取离子信息,用Trace Finder软件建立数据库,数据库包含73种化合物的电离模式、母离子质量数、保留时间、二级特征碎片离子。
2.2 定性及定量
2018年,《食品中那非类物质的测定》食品补充检验方法的公告[25]指出,使用高分辨定性判定需要在试样中检出与某标准品色谱峰(或由标准品建立的谱库)保留时间一致的色谱峰,并且与标准品母离子精确分子量误差不超过5×10-6,主要碎片离子精确分子量误差不超过10-5。从表1可以看出,本研究得到的数据库内容能够满足高通量快速筛查定性的要求。定量方法采用外标法,将目标物母离子峰面积为纵坐标y,以目标物的浓度为横坐标x,绘制标准曲线,根据待测物母离子峰面积进行定量计算。
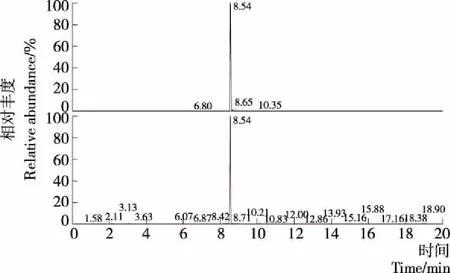
图1 西布曲明和去甲基他达那非提取离子流图Figure 1 Extracted chromatograms of sibutramine and nortadalafil

表1 73种化合物参数Table 1 Parameters of 73 compounds
续表1

化合物名称分子式保留时间扫描模式母离子(m/z)碎片离子(m/z)巴比妥C8H12N2O36.76[M-H]-183.07752140.0717,94.0662苯巴比妥C12H12N2O37.90[M-H]-231.07752188.0717,85.0044司可巴比妥C12H18N2O38.84[M-H]-237.12447194.1187,85.0044异戊巴比妥C11H18N2O38.59[M-H]-225.12447157.0608,111.1168氯美扎酮C11H12ClNO3S8.30[M+H]+274.02992154.0418,209.0602佐匹克隆C17H17ClN6O36.97[M+H]+389.11234245.0225,130.0053氯苯那敏C16H19ClN27.30[M+H]+275.13095230.0731,167.0730扎来普隆C17H15N5O8.54[M+H]+306.13494264.1244,236.0904文法拉辛C17H27NO27.47[M+H]+278.21146215.1430,121.0648青藤碱C19H23NO46.26[M+H]+330.16998181.0648,153.0700罗通定C21H25NO47.48[M+H]+356.18563165.0911,192.1018褪黑素C13H16N2O27.68[M+H]+233.12845174.0908,131.0726奥沙西泮C15H11ClN2O28.65[M+H]+287.05818241.0527,163.0058劳拉西泮C15H10Cl2N2O27.59[M+H]+321.01921289.0294,177.0214尼群地平C18H20N2O610.04[M-H]-359.12486226.0863,210.0913尼莫地平C21H26N2O710.22[M-H]-417.16672301.0819,255.0890尼索地平C20H24N2O610.41[M+H]+389.17073211.0850,183.0912非洛地平C18H19Cl2NO410.63[M-H]-382.06184278.0134,243.0445氢氯噻嗪C7H8ClN3O4S26.32[M-H]-295.95720204.9833,157.0162哌唑嗪C19H21N5O47.13[M+H]+384.16663247.1190,138.0550利血平C33H40N2O98.45[M+H]+609.28066448.1967,397.2122卡托普利C9H15NO3S7.10[M+H]+218.08454172.0792,116.0706可乐定C9H9Cl2N35.86[M+H]+230.02463212.9981,186.9824氨氯地平C20H25ClN2O58.13[M+H]+409.15248238.0629,294.0891硝苯地平C17H18N2O69.39[M+H]+347.12376315.0976,195.0917阿替洛尔C14H22N2O34.64[M+H]+267.17032190.0863,116.1070洛伐他汀C24H36O510.36[M+H]+405.26355199.1481,143.0855辛伐他汀C25H38O511.42[M+H]+419.27920199.1481,173.1325烟酸C6H5NO21.22[M+H]+124.0393078.0338,96.0444洛伐他汀羟酸钠盐C24H37NaO610.37[M-Na]-421.25956319.1902,101.0601美伐他汀C23H34O510.71[M+H]+391.24790155.0841,185.1316脱羟基洛伐他汀C24H34O411.90[M+H]+387.25299131.0862,199.1490那红地那非C24H32N6O37.49[M+H]+453.26087297.1346,113.1073红地那非C25H34N6O37.54[M+H]+467.27652297.1346,127.1230羟基豪莫西地那非C23H32N6O5S7.76[M+H]+505.2227799.0917,283.1190西地那非C22H30N6O4S7.80[M+H]+475.21220311.1503,283.1190氨基他达拉非C21H18N4O48.49[M+H]+391.14008269.1033,262.0863他达拉非C22H19N3O48.87[M+H]+390.14483268.1081,135.0441硫代艾地那非C23H32N6O3S28.75[M+H]+505.20501327.1274,299.0961,去甲基他达拉非C21H17N3O48.54[M+H]+376.12918302.0812,262.0862硫代西地那非C22H30N6O3S28.62[M+H]+491.18936100.0995,341.1424豪莫西地那非C23H32N6O4S7.85[M+H]+489.22785114.1152,311.1503伐地那非C23H32N6O4S7.42[M+H]+489.22785312.1581,151.0866伪伐地那非C22H29N5O4S9.54[M+H]+460.20130169.0974,312.1581那莫西地那非C22H29N5O4S10.55[M+H]+460.20130312.1581,283.1195
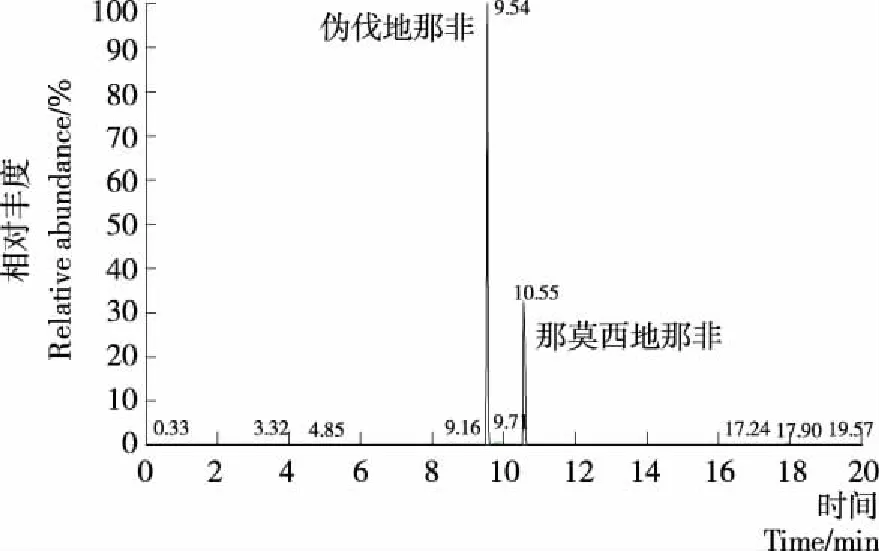
图2 伪伐地那非和那莫西地那非提取离子流图Figure 2 Extracted chromatograms of vardenafil and norneosildenafil
2.3 基质效应
基质效应由分析物的共流出组分影响电喷雾接口的离子化效率所致,表现为离子增强或离子抑制作用,仪器的分辨率及抗干扰能力也影响基质效应,若有较强的基质增强或基质减弱效应的一般采取基质匹配标准曲线进行定量分析。Orbitrap具有超高分辨率,背景噪音干扰极小,基质效应相对较低。为了进一步明确保健食品中的基质效应,本研究验证了片剂、胶囊、口服液的基质效应,均在80%~120%,因此定量时不考虑基质效应带来的影响。
2.4 线性关系及检出限
采用Orbitrap高分辨质谱对73种非法添加的西药进行定量分析,以目标物母离子特征离子峰面积作为纵坐标y,以目标物的浓度为横坐标x,绘制标准曲线,所有化合物在一定的范围内线性关系良好,R>0.99。在空白片剂样品中添加低浓度的目标化合物,检出限(LOD)以S/N=3确定,S/N=10时对应浓度为定量限,各化合物的检出限及定量限见表2。

表2 各化合物检出限及定量限Table 2 The LOD and LOQ of 73 compounds
续表2

化合物名称线性范围/(ng·mL-1)R检出限/(μg·kg-1)定量限/(μg·kg-1)化合物名称线性范围/(ng·mL-1)R检出限/(μg·kg-1)定量限/(μg·kg-1)氯硝西泮0.6~2000.99926羟基豪莫西地那非0.6~2000.99826三唑仑0.6~2000.99926西地那非0.6~2000.99826地西泮0.6~2000.99926氨基他达拉非1.5~3000.999515巴比妥6.0~5000.9982060他达那非0.6~2000.99926苯巴比妥6.0~5000.9982060硫代艾地那非1.5~3000.999515司可巴比妥6.0~5000.9982060去甲基他达拉非0.6~2000.99926异戊巴比妥6.0~5000.9992060硫代西地那非1.5~3000.999515氯美扎酮1.5~3000.998515豪莫西地那非0.6~2000.99926佐匹克隆1.5~3000.999515伐地那非0.6~2000.99926氯苯那敏1.5~3000.998515伪伐地那非0.6~2000.99826扎来普隆1.5~3000.999515那莫西地那非1.5~3000.998515文法拉辛1.5~3000.998515
2.5 回收率及精密度
在胶囊、片剂、口服剂3种保健食品基质中按照100,500,1 000 μg/kg含量添加标准溶液,每个水平重复3次。经计算,其回收率为72.2%~117.5%,相对标准偏差为2.3%~6.8%,表明该方法是一种适合不同基质保健食品非法添加高通量快速筛查定量的方法。
2.6 实际样品检测
利用本方法对市售28份保健食品进行检测,1批次检出洛伐他汀,1批次检出他达那非。
3 结论
本研究采用甲醇超声提取,建立了保健食品中非法添加西药成分高通量筛查及定量方法,该方法简单、快速,精密度及准确性高,不仅可以作为快速筛查及定量73种化合物的方法,还可以根据检测需求反复调用,进行其他目标化合物的靶向确证及非靶向筛查,扩宽了数据的可利用性,但本研究的非法添加药物种类仍然有限,今后应不断完善补充数据库。
猜你喜欢
化工设计通讯(2022年10期)2022-12-31
波谱学杂志(2022年2期)2022-06-14
中国现代医药杂志(2020年12期)2020-02-06
化工设计通讯(2020年11期)2020-01-12
中成药(2018年12期)2018-12-29
中成药(2018年12期)2018-12-29
基层中医药(2018年7期)2018-12-06
知识经济·中国直销(2018年11期)2018-11-26
中国当代医药(2015年36期)2015-03-11
中国卫生(2014年2期)2014-11-12
