
绿豆SSR标记的开发及遗传多样性分析
2019-07-23 08:40:36叶卫军陈圣男张丽亚田东丰
作物学报 2019年8期
叶卫军 陈圣男 杨 勇 张丽亚 田东丰 张 磊 周 斌
绿豆SSR标记的开发及遗传多样性分析
叶卫军**陈圣男**杨 勇 张丽亚 田东丰 张 磊 周 斌*
安徽省农业科学院作物研究所, 安徽合肥 230031
SSR标记以其数量丰富、多态性好、共显性遗传等优点在基础研究和育种工作中发挥了重要作用, 但目前绿豆基因组中的SSR标记依然较少。本研究将磁珠富集法和测序技术相结合高通量检测绿豆基因组SSR位点, 鉴定出3,275,355个SSR位点, 开发了2742个SSR标记。选取其中157个SSR进行PCR验证, 发现有90个(57.33%)标记在10份材料中表现出多态性。挑选40个条带清晰、多态性高、染色体上均匀分布的标记对90份绿豆资源进行遗传多样性分析, 单个位点检测到的等位变异数为2~8个, 平均为3.0个, 有效等位基因数为1.31~4.21个, 平均为2.16。Nei’s基因多样性指数在0.23~0.76之间, 平均为0.51。多态性信息含量为0.22~0.72, 平均为0.43。聚类分析将90份材料分为2个类群, 包含4个组。第I组主要由北方资源组成, 第II组种质来源较为分散, 第III组主要由山东的资源构成, 第IV组包含多数河北的种质资源。本研究开发的多态性SSR标记不仅可以用于绿豆种质资源的遗传多样性分析, 也将在高密度遗传图谱构建、基因定位和分子标记辅助育种中发挥重要作用。
绿豆; 测序; SSR; 引物设计; 遗传多样性
绿豆([L.] Wilczek), 别名青小豆、菉豆、植豆, 是豆科(Leguminosae)蝶形花亚科(Papilionaceae)菜豆族(Phaseoleae)豇豆属()的1个栽培种, 染色体数2= 2= 22。绿豆是喜温作物, 起源于中国, 已有2000多年的种植历史[1]。但也有证据表明绿豆原产于印度, 约在3500年前被驯化[2]。目前, 绿豆主要在温带、亚热带和热带地区种植, 年种植面积600万公顷[3]。绿豆属高蛋白、中淀粉、低脂肪类食物, 富含多种矿质元素、维生素和活性物质, 具有抗菌、抗肿瘤、降血压和解毒作用, 一直作为药食同源作物[4]。绿豆适应性广, 生育期短, 抗逆性强, 并具有共生固氮、改良土壤的能力, 可与玉米、棉花、薯类等作物间套种, 提高经济效益[5-6]。但长期以来, 绿豆一直被视作小杂粮, 基础研究水平远落后于水稻、小麦和玉米等大宗作物。Kang等[7]2014年通过测序技术得到大小为431 Mb的绿豆基因组草图, 为绿豆遗传育种和功能基因组学的发展奠定了基础。
SSR (simple sequence repeat)标记因其共显性、多态性高、易操作、耗费低等优点, 在绿豆遗传多样性分析[8-9]、遗传连锁图谱构建[10-11]和QTL定位[12]等方面的应用越来越广泛。而且有研究报道SSR标记在豇豆属作物中也有一定的通用性[13-14]。由于绿豆分子标记的研究起步较晚, 现已发布的SSR标记数量有限[15-21]。随着测序技术的发展, 富集文库和高通量测序成为目前常用的SSR位点的鉴定方法。Wang等[22]通过构建SSR-富集文库的方法开发了6100个SSR标记, 发现只有9.10%的引物在野生资源和栽培种中表现出多态性。选取的1700对引物中仅有49对(2.88%)在32份绿豆资源中表现出多态性, 说明这些引物的多态性水平较低。Chen等[23]利用高质量的转录组数据发现了13,134个EST-SSR位点, 对其中200个位点的验证发现, 有66个(33.00%)标记在31份资源中表现出多态性。Liu等[24]利用转录组数据, 鉴定出3788个EST-SSR位点, 其中320个位点中有53个(16.56%)在4份栽培品种中表现出多态性。由于这些研究利用的是单个品种的序列信息, 引物在多份种质中的多态性结果无法分析, 标记开发后需大量的引物验证工作, 且多态性标记数量较少, 严重限制了SSR标记的使用效率。本研究利用磁珠富集和高通量测序相结合的方法高效获取多个品种的重复序列片段信息, 通过序列聚类和SSR位点长度多态性分析进行SSR引物的设计, 提高了标记开发的效率。
1 材料与方法
1.1 试验材料
研究材料为本单位从全国各地收集、整理、保存的90份绿豆资源(表1), 包括北京12份、安徽12份、河南4份、江苏11份、吉林6份、河北17份、湖北2份、内蒙古1份、辽宁2份、黑龙江1份、山西12份、山东8份、重庆2份。选取中绿5号、苏绿2号、皖科绿3号、潍绿11号、潍绿12号等10份地理来源不同且表型差异较大的绿豆品种用于SSR引物的开发及多态性验证。
1.2 基因组文库构建与探针设计
采用植物基因组DNA提取试剂盒(天根生化科技有限公司)提取DNA, 将测序所用的10个DNA样本等质量混合后随机片段化构建DNA文库, 文库的插入片段大小控制在400 bp左右。SSR富集所采用的探针包括p(AG)10、p(AC)10、p(AAC)8、p(ACG)8、p(AAG)8、p(AGG)8、p(ACAT)6和p(ATCT)68种。具体操作步骤参考孙子奎等[25]的方法。
1.3 数据获取、整理与SSR位点发掘
利用Illumina MiSeq2000平台测序, 测序模式为Paired-end (双端)。测序完成后, 采用Adapter Removal V2.1.7[26]去除接头污染, 并删除测序质量较差和较短的序列。利用FLASH V1.2.11[27]软件对双端测序的序列进行整合。用微卫星识别工具(Microsatellite identification tool, MISA, http://pgrc. ipkgatersleben.de/misa/)从所有序列中查找SSR位点。SSR位点的筛选标准为单核苷酸、二核苷酸、三核苷酸、四核苷酸、五核苷酸和六核苷酸的重复次数最少为10、6、5、5、5和5次。
1.4 序列聚类及SSR多态性评估
采用Perl程序屏蔽序列上的重复序列, 过滤掉侧翼序列短于20 bp的短序列, 并利用cdhit软件对过滤后的核苷酸序列进行聚类, 聚类所采用的核苷酸序列相似度设置为95%。若一条序列上有2个及以上SSR的序列则分开统计聚类群组。用Perl程序解析聚类结果, 根据SSR的长度分别统计每一类, 若同一类中所有SSR的长度一致, 则该类的多态性为1; 如果同一类中SSR具有2种长度, 则该类的多态性为2; 依此类推, 获得每一类SSR长度的多态性数据。
1.5 引物设计与筛选
用Primer3 V2.3.6[28]对群组内多态性≥2的SSR序列的两端设计引物。选取SSR类型为非单核苷酸重复单元、非复合重复单元且每条序列上只有1个SSR的序列设计引物。引物的物理位置和大小参考绿豆基因组序列(https://www.ncbi.nlm.nih.gov/ genome/664)。筛选后的引物用PCR验证多态性, 反应体系包含50 ng μL-1基因组DNA 1 μL、正反向引物(10 μmol L-1)各0.5 μL、2×PCR Master Mix (TIANGEN) 5.0 μL, 补水至10 μL。PCR扩增程序为94℃预变性5 min; 94℃变性30 s,m(根据引物设定) 30 s, 72℃延伸2 min, 35个循环; 72℃延伸10 min, 12℃保存。将PCR产物用8%的聚丙烯酰胺电泳, 银染后读带。
表1 供试绿豆材料信息
1.6 遗传多样性分析
按照各SSR位点PCR扩增片段迁移率的不同分别读取数据, 并依据分析软件的要求相应转换数据格式。用POPGENE V1.32[29]软件计算等位基因数(allele number, Na)和有效等位基因数(effective number of allele, Ne)。用PowerMarker V3.25软件[30]计算标记多态性信息含量(polymorphism information content, PIC)和Nei’s基因多样性(Nei’s gene diversity, H)。基于Nei’s遗传距离并采用UPGMA (unweight pair group method using arithmetic averages, 非加权平均数)法对供试绿豆资源进行聚类分析。
2 结果与分析
2.1 测序数据整理
采用双末端测序的方法对SSR富集文库进行测序, 共得到原始测序序列11,105,850条, 包含3,253,148,219 bp。过滤数据, 去除接头污染和低质量的测序序列得到高质量的序列10,413,002条, 占原始测序序列总数的93.76%, 包含2,474,490,321 bp, 占测序总碱基数的76.06%。过滤后的数据Q20 (碱基识别准确率在99%以上的碱基所占百分比)和Q30 (碱基识别准确率在99.9%以上的碱基所占百分比)分别为98.54%和92.99%, 表明该文库的测序质量较好, 可满足后续分析的需求。
2.2 SSR搜索与多态性评估
采用FLASH V1.2.11对过滤后序列的R1端和R2端进行整合。可以合并的序列数为4,577,639对, 占总数(5,206,501)的87.92%, 合并后序列总长度为1,582,586,848 bp。用SSR识别工具在所有整合后的序列中搜索SSR位点, 共得到3,275,355个SSR位点, 频率为71.55%, 平均每0.48 kb一个SSR位点。这些SSR位点分布在1,146,445条序列中, 其中有705,057条(61.50%)序列包含1个以上SSR, 以复合形式存在的SSR数量为2,077,761个(63.44%)。对这些位点分析发现, 单、二、三、四、五、六核苷酸重复单元的数量分别有337,267、343,776、2,580,413、8624、3429和1846个, 分别占总数的10.30%、10.50%、78.78%、0.26%、0.10%和0.06% (图1-A)。三核苷酸重复单元中AAC/GTT类型有2,341,675个, 占该类型的90.75% (图1-D), 而CCG/CGG型出现频率最低, 只有60个。单核苷酸重复单元中, A/T类型的重复序列出现频率最多, 占该类型的98.94%, G/C类型仅有3584个, 占1.06% (图1-B)。二核苷酸重复单元有4种类型, AC/GT类型有209,412个, 占60.84%, 为主导地位, 其次为AT/AT和AG/CT型, 最少的为CG/CG型, 只有75个(图1-C)。四核苷酸重复单元出现22种类型, 其中ACAT/ATGT和AAAT/ATTT出现频率较高, 分别为45.55%和29.20%, 其他几种重复类型的出现频率均低于0.1% (图1-E)。五核苷酸重复类型共有36种, 其中AAAAT/ATTTT和AAATC/ATTTG出现频率较高, 分别为47.94%和29.54% (图1-F)。六核苷酸重复类型有74种, 出现频率较高的为AACAAG/CTTGTT和AAGAGG/CCTCTT, 占比29.14%和11.65% (图1-G)。
2.3 SSR引物设计
对含有SSR位点的序列进行过滤, 侧翼序列长度超过20 bp的共有661,715条。对过滤后的序列按序列相似度进行聚类, 发现这些序列分布在534,209个类群中。根据SSR的长度分别对每一类统计分析, 有92.15%的SSR位点在选取的10份材料中不表现出长度多态性, 剩余的SSR多数长度多态性为2, 表现出高多态性的SSR只占较少比例(表2)。利用引物设计软件对SSR长度多态性≥2的序列两端设计引物, 得到2742对引物。
2.4 SSR引物筛选与多态性分析
对设计的引物按SSR长度多态性高、染色体上均匀分布等原则进行筛选后, 选取157对引物进行多态性验证。有90对引物可扩增出多态性片段, 占比57.33% (表3), 其余有15对引物表现出非特异性扩增或无扩增产物、52对引物可扩增出目的条带但无多态性(图2-A)。同时发现, 各染色体上的多态性引物比率差异较大, 第1、第3、第4染色体上的多态性引物比率较高, 分别有10、9和7个, 占比均超过了70%, 而第9染色体上的引物多态性比率最低, 只有4个, 只占29.57% (图2-B)。
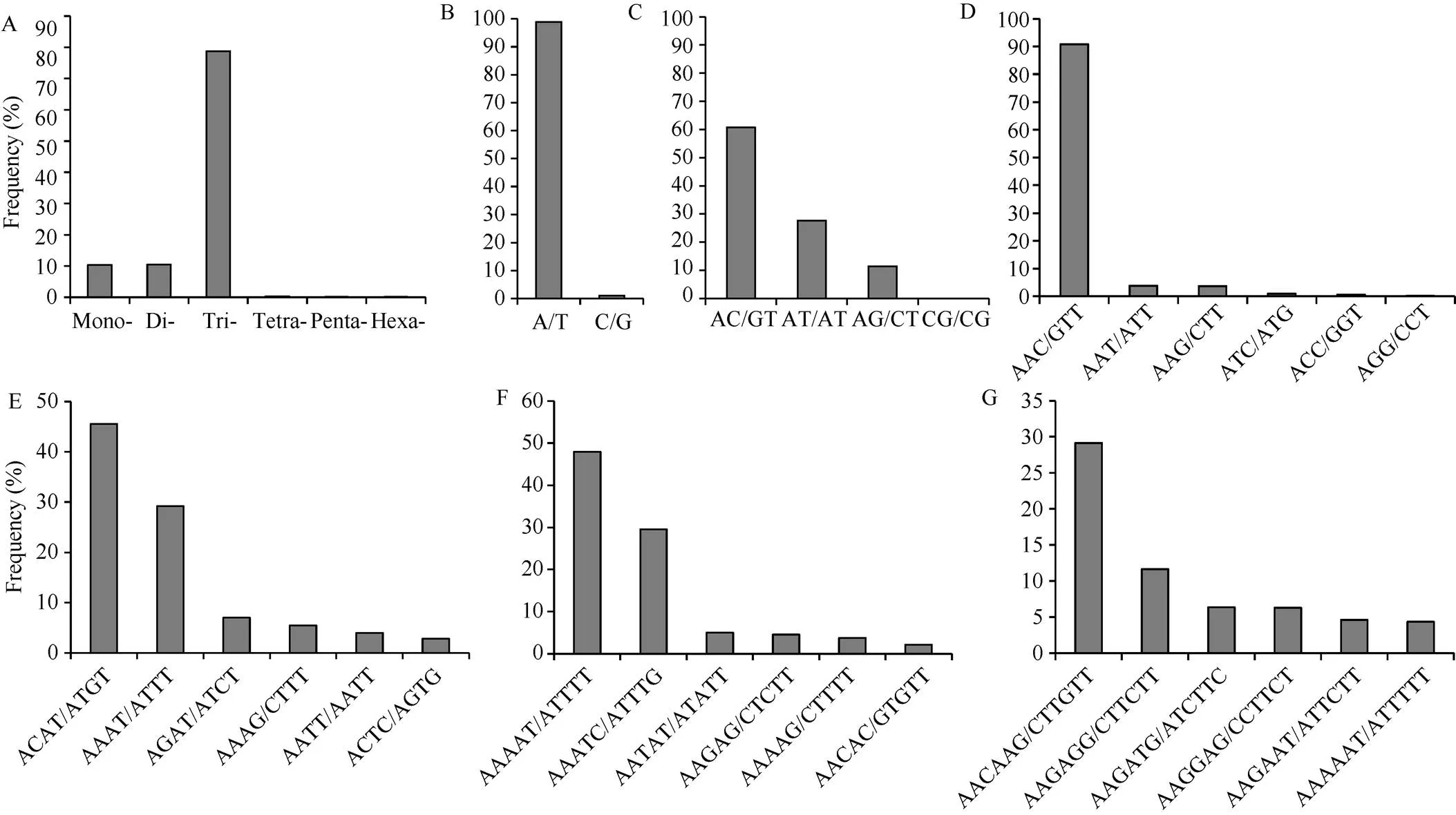
图1 SSR的类型和频率分布
A: 不同类型SSR的分布频率, Momo-、Di-、Tri-、Tetra-、Penta-、Hexa-分别代表单核苷酸、二核苷酸、三核苷酸、四核苷酸、五核苷酸和六核苷酸重复; B~G: 单核苷酸(B)、二核苷酸(C)、三核苷酸(D)、四核苷酸(E)、五核苷酸(F)和六核苷酸(G)的重复类型及频率。
A: frequency of different SSR types. Momo-, Di-, Tri-, Tetra-, Penta-, and Hexa-represents mononucleotide, dinucleotide, trinucleotide, tetranucleotide, pentanucleotide and hexanucleotiede repeat motif, respectively. B-G: Mon-(B), Di-(C), Tri-(D), Tetra-(E), Penta-(F), and Hexa-(G) motif types and frequency.
2.5 SSR引物特征分析
从90对引物中选取40对条带清晰且多态性较高的引物对90份绿豆资源进行遗传多样性分析, 引物信息见表4。可以看出, 单个引物检测到的等位基因数在2~8个, 平均为3个, 等位基因数最高的为Vr11-4, 有8个。有效等位基因数为1.31~4.21个, 平均为2.16个。Nei’s基因多样性指数在0.23 (Vr8-4)~ 0.76 (Vr3-2)之间, 平均为0.51。引物多态性信息含量在0.22~0.72之间, 平均为0.43。PIC小于0.25的低多态性引物仅有1个(Vr8-4), PIC大于0.5高多态性位点有13个, 占比32.5%。
2.6 绿豆种质资源的聚类分析
基于UPGMA聚类分析, 90份材料可分为A、B两个大类群, 包含I、II、III、IV 4个组(图3)。第I组包含21份种质, 分别来源于安徽(5份)、北京(2份)、河北(2份)、吉林(4份)、辽宁(1份)、内蒙古(1份)、山西(6份), 除安徽种质外, 其余为东北和华北资源。第II组包含22份种质, 分别来源于安徽(3份)、江苏(6份)、山东(1份)、北京(6份)、黑龙江(1份)、吉林(1份)、湖北(1份)、山西(3份), 种质地理分布较为分散。第III组包含10份种质, 分别来源于河南(1份)、山东(7份)、北京(1份)和辽宁(1份), 主要为山东种质。第IV组包含31份种质, 分别来源于安徽(2份)、北京(2份)、重庆(2份)、江苏(5份)、河南(3份)、河北(14份)和山西(3份), 河北种质主要聚类在该组。可见具有相同地理来源的材料多数可以聚在一起或成簇状分布, 表明来源相同的种质具有较近的亲缘关系。
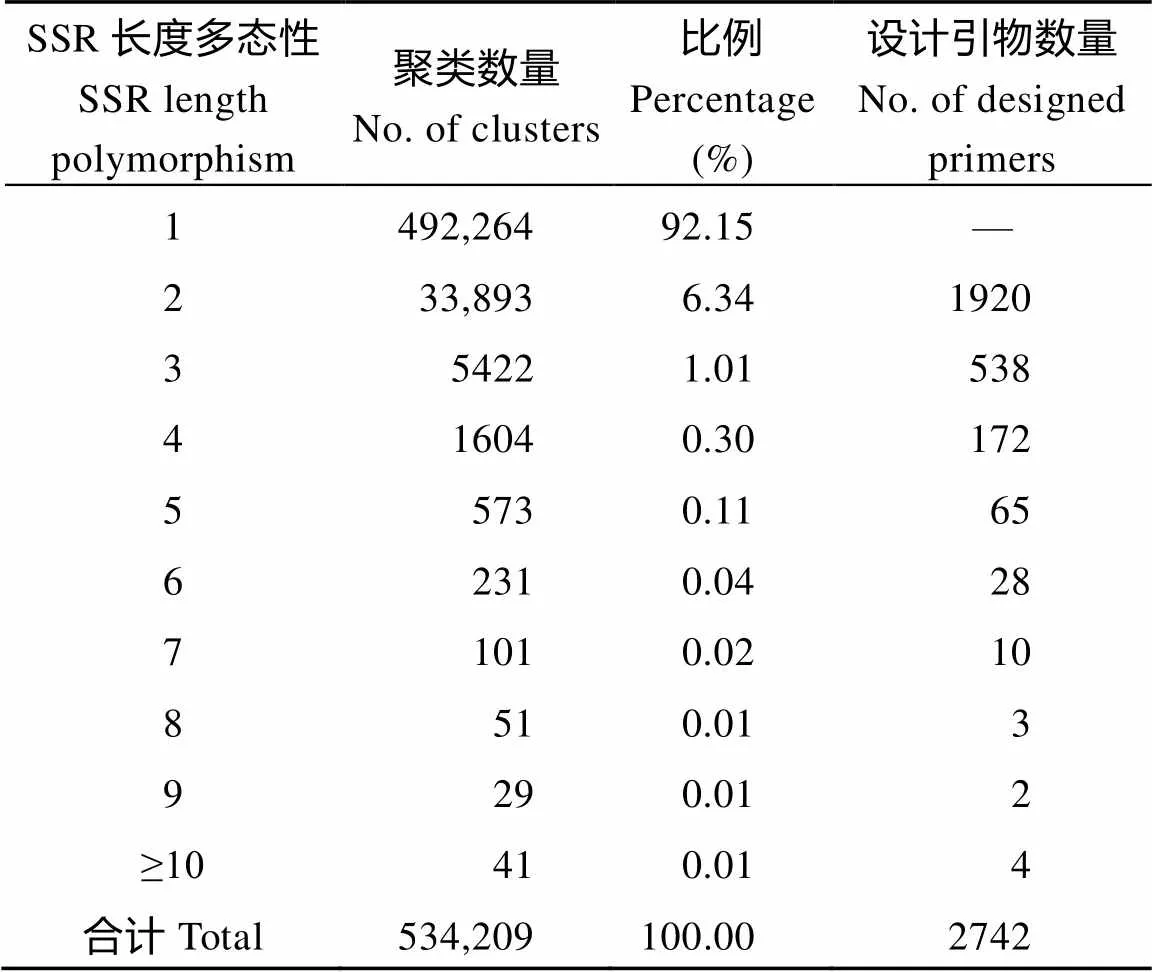
表2 SSR长度多态性评估
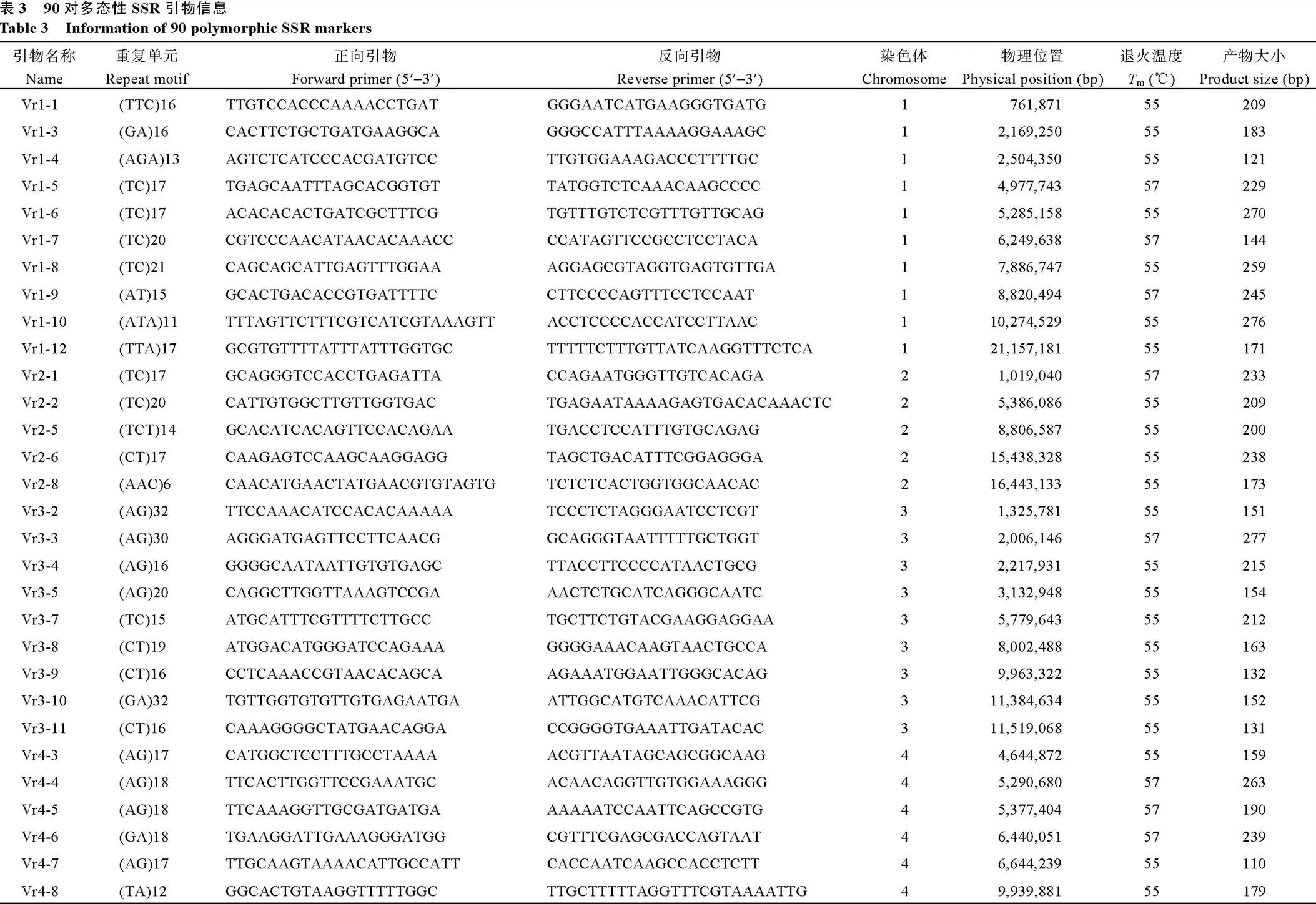
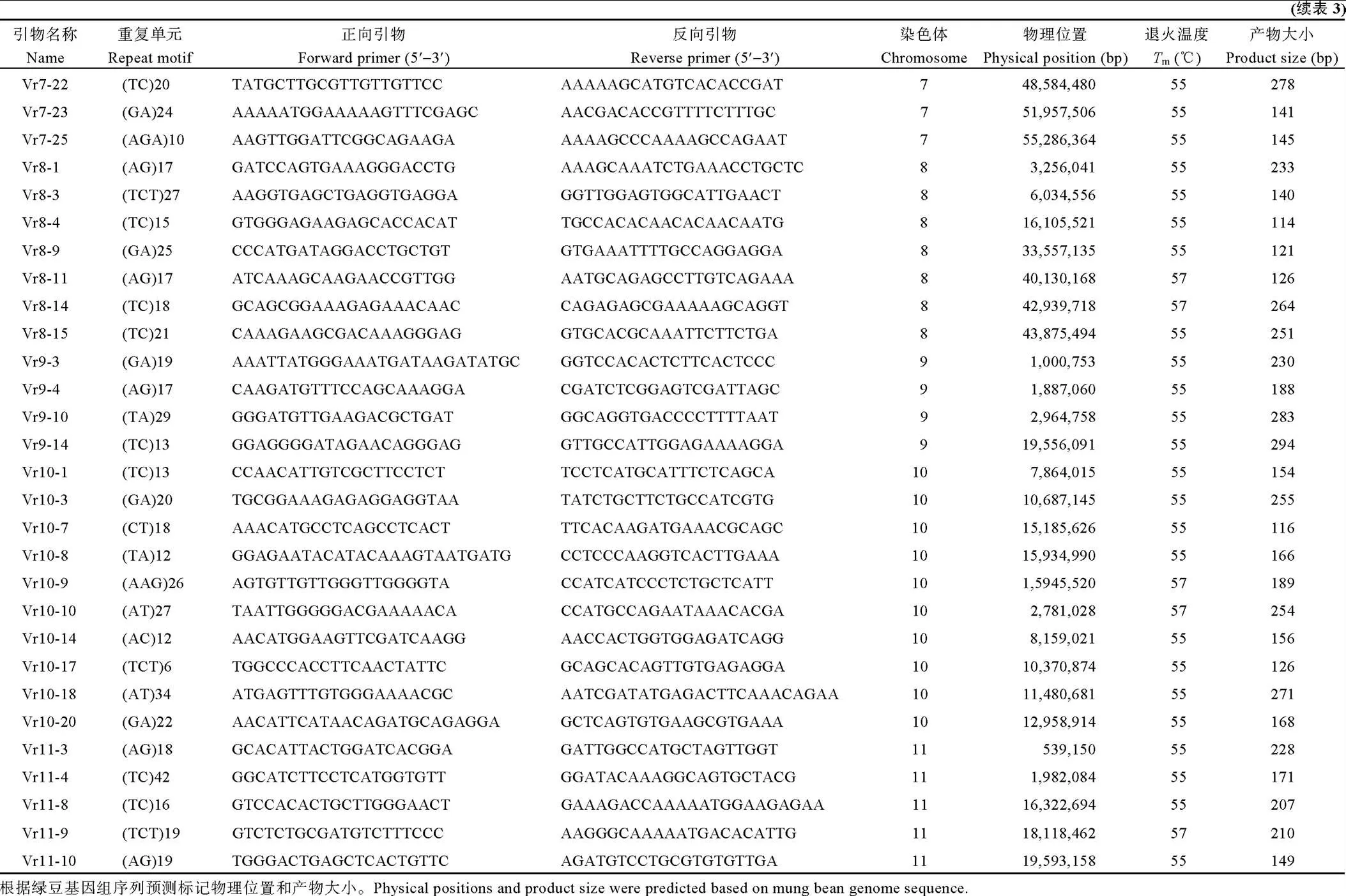
A: 157对引物PCR结果的统计分析; B: 11条染色体上的引物扩增情况。MSP表示多态性扩增的标记, MSN表示可扩增出目的条带但无多态性的标记, MNN表示非特异性扩增或无扩增产物的标记。
A: statistical analysis of PCR amplification results of the 157 primers; B: primer amplification results on the 11 chromosomes. MSP: markers amplified specific and polymorphic bands; MSN: markers amplified specific and non-polymorphic bands; MNN: markers amplified non-specific or no bands.
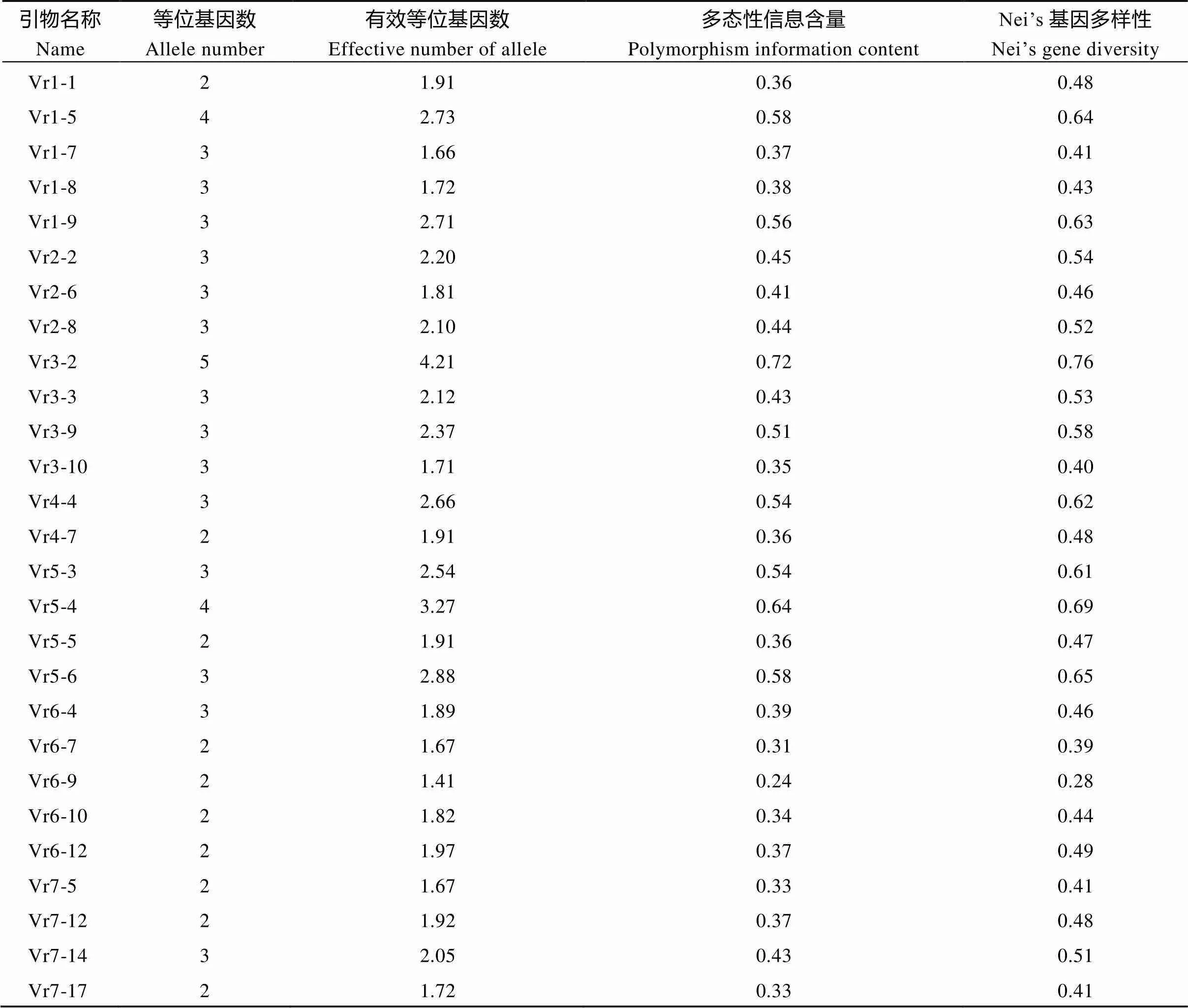
表4 40对SSR引物的信息
(续表4)
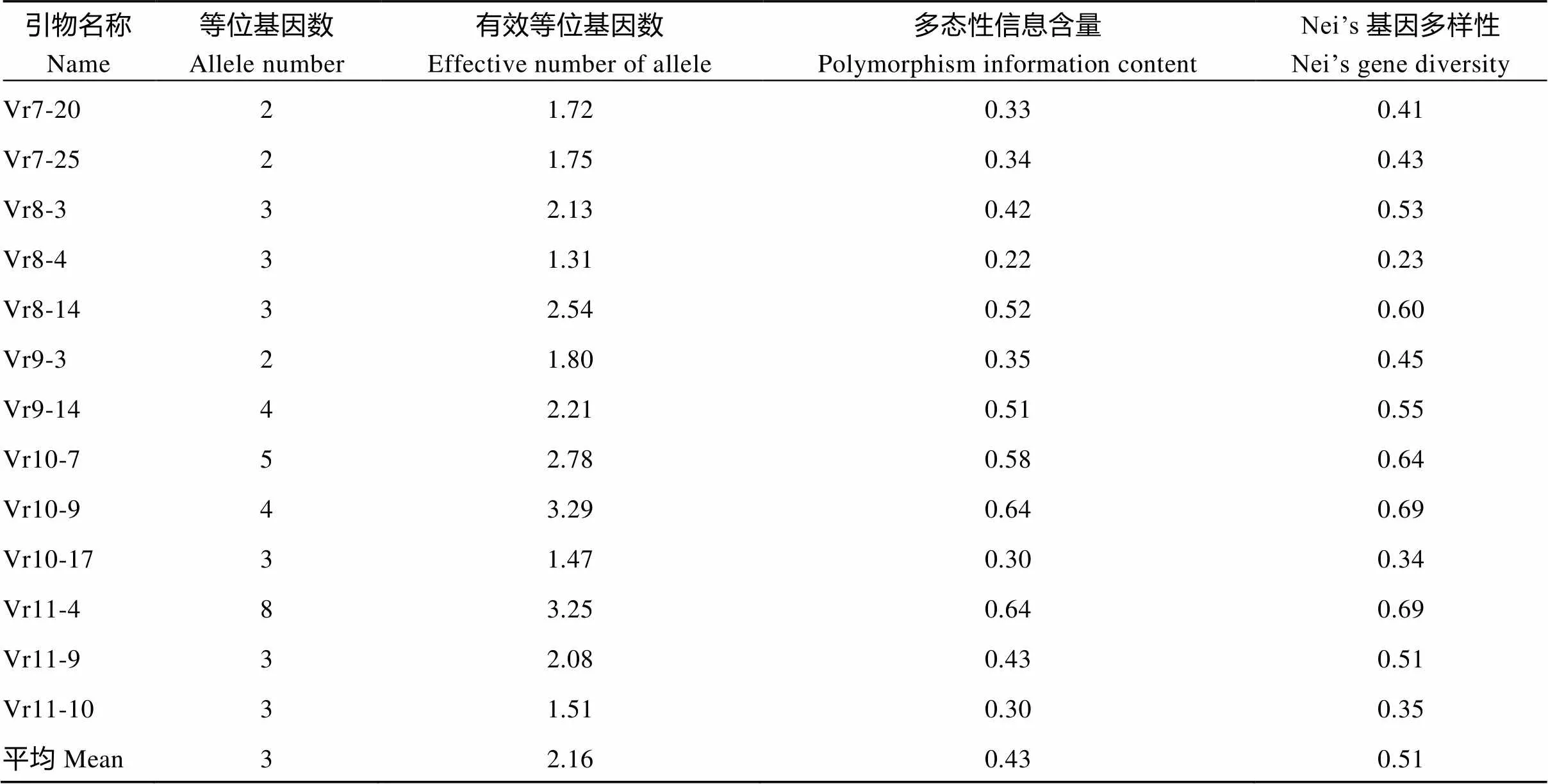
引物名称Name等位基因数Allele number有效等位基因数Effective number of allele多态性信息含量Polymorphism information contentNei’s基因多样性Nei’s gene diversity Vr7-2021.720.330.41 Vr7-2521.750.340.43 Vr8-332.130.420.53 Vr8-431.310.220.23 Vr8-1432.540.520.60 Vr9-321.800.350.45 Vr9-1442.210.510.55 Vr10-752.780.580.64 Vr10-943.290.640.69 Vr10-1731.470.300.34 Vr11-483.250.640.69 Vr11-932.080.430.51 Vr11-1031.510.300.35 平均Mean32.160.430.51
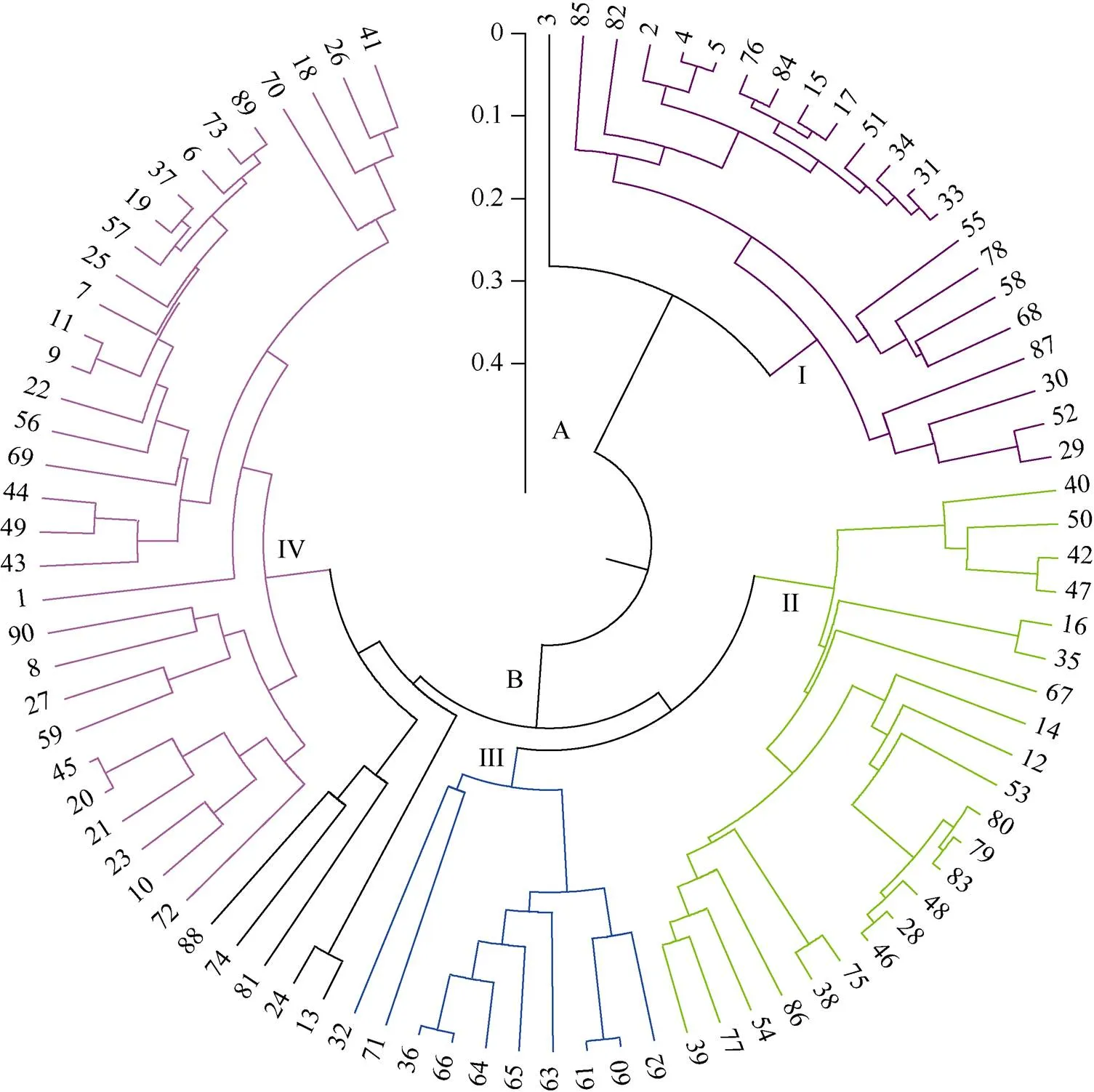
图3 根据Nei’s遗传距离的绿豆资源UPGMA聚类图
1~90为表1中供试绿豆编号。
1-90 correspond with the number of mung bean accessions given in Table 1.
3 讨论
目前, 开发SSR标记的方法有很多种。传统的磁珠富集法优势在于可高效获得具有重复序列的片段, 但需要挑选阳性克隆测序后才能设计引物, 费时费力, 且多态性引物比率不高。吴传书[31]利用磁珠富集法开发6100对绿豆SSR引物, 只有559对在24份绿豆材料中表现出多态性, 多态率仅为9.16%。采用生物信息学技术开发SSR标记也是目前常用的方法, 其优点在于可以获取大量的序列信息, 便于高通量操作。但该方法需要对基因组或转录组测序后才能进行SSR位点的检索, 比较适用于基因组序列信息较丰富的物种。且这种方法获取的数据多为单个品种的序列信息, 无法对SSR位点在多份资源中的多态性进行评估, 引物的多态率也较低, 尤其不适用于绿豆等遗传多样性较低的物种。本研究将磁珠富集法和测序技术的优势结合起来, 高通量获得多个品种的重复序列片段序列信息, 平均每0.48 kb就有1个SSR位点, 高于Tangphatsornruang等[20]鉴定的1/67 kb的频率和Gupta等[32]发现的1/3.4 kb的频率。在鉴定的SSR中, 以复合形式存在的SSR占据较高的比例(63.44%)。Wang等[22]也发现绿豆基因组中有43.10%的SSR以复合形式存在, 但从绿豆EST序列中发现的复合SSR占比却非常低[23,32], 这表明复合SSR可能多数存在于非编码区。在所有的SSR位点中, 三核苷酸重复单元为主导类型, 与Gupta等[32]和Wang等[22]的结果一致, 但与Tangphatsornruang等[20]和Chen等[23]的结果有偏离。其次为二核苷酸和单核苷酸重复, 而四核苷酸、五核苷酸和六核苷酸重复只占总数的0.42%。在三核苷酸重复单元中, AAC/GTT型占该类型的比例高达90.75%, 与Wang等[22]结果一致, 且与蚕豆中的SSR鉴定结果类似[33]。进一步分析发现C/G (CG/CG或CCG/CGG)型SSR位点出现的频率均较低, 这在前人研究中也有报道。Wang等[22]鉴定的单核苷酸重复类型中A/T占比为88.30%, Chen等[23]和Liu等[24]发现A/T型占单核苷酸重复的比例高达99.70%和95.71%, 且Liu等[24]在二核苷酸重复单元中也仅发现2个CG/GC位点。在其他作物如大豆[34]和小麦[35]中也发现有类似情况。这可能由于富含C/G的序列主要存在于基因组的编码区, 也可能与绿豆基因组具有较低的GC含量(34.30%)有关。
通过对多个品种的相似序列聚类后, 将SSR位点在各品种中的长度进行多态性分析。发现92.15%的SSR长度多态性为1, 这与绿豆栽培资源遗传变异水平较低有关, 也揭示了前人研究报道中绿豆多态性SSR标记比率较低的原因。采用引物设计软件对长度多态性≥2的位点两端设计引物, 既获得丰富的引物信息, 也提高了多态性引物的比率, 引物验证结果表明多态性引物占总数的57.33%, 远高于Wang等[22]和Chen等[23]的研究结果, 但低于Gupta等[32]的结果。理论上通过这种方法设计的引物应全部表现出多态性, 但由于测序技术和引物设计水平的限制, 部分引物不具有多态性, 在以后的研究中应努力改善。进一步研究发现多态性引物在绿豆基因组上表现为不均匀分布, 部分染色体区域上的多态性引物较少, 这可能与引物所在的基因组位置(如端粒、着丝粒、基因组保守区域)有关, 在以后的引物筛选中应加强这部分染色体区域的引物设计。
SSR标记作为分子标记的一种, 可在DNA水平上揭示材料的遗传变异, 且不受植株发育时期和地理条件限制, 具有其他形态学标记或生理生化标记不可比拟的优点。利用分子标记研究绿豆资源遗传多样性已有多篇报道, 但通常是随机选择一些多态性引物, 引物在染色体上分布可能不均匀, 不能从基因组整体水平上反应资源的遗传变异[8-9]。本研究挑选40对条带清晰、多态性较好且在染色体上均匀分布的引物对绿豆资源进行遗传多样性分析, 能够从基因组整体水平上揭示资源的遗传变异, 结果更为准确可靠。引物检测到的等位变异数在2~8个, 平均为3个, 与王丽侠等[13]和刘岩等[8]的研究结果基本一致。引物PIC值在0.22~0.72之间, 平均为0.43, 高于前人的研究结果[8-9,13], 表明选取的引物适用于资源遗传多样性分析。聚类结果显示相同地理来源的材料多数可以聚在一起, 表明来源相同的种质具有较近的亲缘关系, 但也揭示了该地区绿豆资源遗传基础较为狭窄, 在以后的育种工作中应加强外来种质的引进和使用, 提高品种的遗传多样性。同时也发现部分种质没有严格按照地理来源聚类, 尤其是来源于北京的中绿系列和来源于江苏的苏绿系列的种质在聚类图上分布较为分散。这可能与所用引物及材料的数量和来源有关, 也可能是因为这部分绿豆资源作为亲本在国内各地区间频繁交换使用。本研究所用种质多来源于东北、华北、华东等绿豆主产区, 南方绿豆资源数量不够丰富, 因此不能全面体现国内绿豆种质的遗传变异水平, 在以后的研究工作中应加强南方绿豆资源的收集和遗传多样性分析。
4 结论
以磁珠富集法结合测序技术高通量检测绿豆SSR位点, 共鉴定出3,275,355个SSR位点, 设计了2742个标记。选取157个引物验证发现, 有57.33%的引物在10份材料中表现出多态性, 多态性引物比率较高。聚类分析将90份材料分为2个大类群, 包含4个组。地理来源较近的资源多数可被聚在一起或成簇状分布, 表明种质地理来源与其亲缘关系的一致性。本研究开发的分子标记适用于绿豆遗传连锁图谱和指纹图谱的构建、基因挖掘和遗传多样性分析等工作。种质资源遗传多样性分析的结果也为绿豆起源与进化、核心种质库的构建、优异种质资源的保护和提高优良材料的利用率奠定基础。
[1] 程须珍, 王述民. 中国食用豆类品种志. 北京: 中国农业科学技术出版社, 2009. pp 19–20.Cheng X Z, Wang S M. Chinese Legumes Variety Pictorial. Beijing: China Agricultural Science and Technology Press, 2009. pp 19–20 (in Chinese).
[2] Fuller D Q, Harvey E L. The archaeobotany of Indian pulses: identification, processing and evidence for cultivation., 2006, 11: 219–246.
[3] Nair R M, Schafleitner R, Kenyon L, Srinivasan R, Easdown W, Ebert A W, Hanson P. Genetic improvement of mung bean., 2012, 44: 177–190.
[4] 郑卓杰, 王述民, 宗绪晓. 中国食用豆类学. 北京: 中国农业出版社, 1997. pp 3–6. Zheng Z J, Wang S M, Zong X X. Food Legumes in China. Beijing: China Agriculture Press, 1997. pp 3–6 (in Chinese).
[5] 王丽侠, 程须珍, 王素华. 绿豆种质资源、育种及遗传研究进展. 中国农业科学, 2009, 42: 1519–1527. Wang L X, Cheng X Z, Wang S H. Advances in research on genetic resources, breeding and genetics of mung bean (L.)., 2009, 42: 1519–1527 (in Chinese with English abstract).
[6] 程须珍, 王素华. 中国绿豆产业发展及科技应用. 北京: 中国农业科学技术出版社, 2002. pp 3–8. Cheng X Z, Wang S H. Indusdustrial Development and Technology Utilization of Mungbean in China. Beijing: China Agricultural Science and Technology Press, 2002. pp 3–8 (in Chinese).
[7] Kang Y J, Kim S K, Kim M Y, Lestari P, Kim K H, Ha B K, Jun T H, Hwang W J, Lee T, Lee J, Shim S, Yoon M Y, Jang Y E, Han K S, Taeprayoon P, Yoon N, Somta P, Tanya P, Kim K S, Gwag J G, Moon J K, Lee Y H, Park B S, Bombarely A, Doyle J J, Jackson S A, Schafleitner R, Srinives P, Varshney R K, Lee S H. Genome sequence of mung bean and insights into evolution within Vigna species., 2014, 5: 5443, doi: 10.1038/ ncomms6443.
[8] 刘岩, 程须珍, 王丽侠, 王素华, 白鹏, 吴传书. 基于SSR标记的中国绿豆种质资源遗传多样性研究. 中国农业科学, 2013, 46: 4197–4209. Liu Y, Cheng X Z, Wang L X, Wang S H, Bai P, Wu C S. Genetic diversity research of mungbean germplasm resources by SSR markers in China., 2013, 46: 4197–4209 (in Chinese with English abstract).
[9] 任红晓, 程须珍, 徐东旭, 高运青, 尚启兵. 应用SSR标记分析中国北方名优绿豆的遗传多样性. 植物遗传资源学报, 2015, 16: 395–399. Ren H X, Cheng X Z, Xu D X, Gao Y Q, Shang Q B. Genetic diversity of traditional mungbean varieties in northern China by SSR markers., 2015, 16: 395–399 (in Chinese with English abstract).
[10] 赵丹, 程须珍, 王丽侠, 王素华, 马燕玲. 绿豆遗传连锁图谱的整合. 作物学报, 2010, 36: 932–939. Zhao D, Cheng X Z, Wang L X, Wang S H, Ma Y L. Integration of mungbean () genetic linkage map., 2010, 36: 932–939 (in Chinese with English abstract).
[11] Isemura T, Kaga A, Tabata S, Somta P, Srinives P, Shimizu T, Jo U, Vaughan D A, Tomooka N. Construction of a genetic linkage map and genetic analysis of domestication related traits in mungbean ()., 2012, 7: e41304.
[12] 王建花, 张耀文, 程须珍, 王丽侠. 绿豆分子遗传图谱构建及若干农艺性状的QTL定位分析. 作物学报, 2017, 43: 1096–1102. Wang J H, Zhang Y W, Cheng X Z, Wang L X. Construction of genetic map and identification of QTLs related to agronomic traits in mung bean., 2017, 43: 1096–1102 (in Chinese with English abstract).
[13] 王丽侠, 程须珍, 王素华, 刘长友, 梁辉. 小豆SSR引物在绿豆基因组中的通用性分析. 作物学报, 2009, 35: 816–820. Wang L X, Cheng X Z, Wang S H, Liu C Y, Liang H. Transferability of SSR from adzuki bean to mungbean., 2009, 35: 816–820 (in Chinese with English abstract).
[14] 钟敏, 程须珍, 王丽侠, 王素华, 王小宝. 绿豆基因组SSR引物在豇豆属作物中的通用性. 作物学报, 2012, 38: 223–230. Zhong M, Cheng X Z, Wang L X, Wang S H, Wang X B. Transferability of mungbean genomic-SSR markers in other vigna species., 2012, 38: 223–230 (in Chinese with English abstract).
[15] Kumar S V, Tan S G, Quah S C, Yusoff K. Isolation and characterization of seven tetranucleotide microsatellite loci in mungbean,., 2002,2: 293–295.
[16] Miyagi M, Humphry M, Ma Z Y, Lambrides C J, Bateson M, Liu C J. Construction of bacterial artificial chromosome libraries and their application in developing PCR-based markers closely linked to a major locus conditioning bruchid resistance in mung bean (L. Wilczek)., 2004, 110: 151–156.
[17] Gwag J G, Chung J W, Chung H K, Lee J H, Ma K H. Characterization of new microsatellite markers in mungbean,(L.)., 2006, 6: 1132–1134.
[18] Somta P, Musch W, Kongsamai B, Chanprame S, Nakasatien S, Toojinda T, Sorajjapinun W, Seehalak W, Tragoonrung S, Srinives P. New microsatellite markers isolated from mungbean ((L.) Wilczek)., 2008, 8: 1155– 1157.
[19] Seehalak W, Somta P, Sommanas W, Srinives P. Microsatellite markers for mungbean developed from sequence database., 2009, 9: 862–864.
[20] Tangphatsornruang S, Somta P, Uthaipaisanwong P, Chanprasert J, Sangsrakru D, Seehalak W, Sommanas W, Tragoonrung S, Srinives P. Characterization of microsatellites and gene contents from genome shotgun sequences of mungbean ((L.) Wilczek)., 2009, 9: 137, doi: 10.1186/1471- 2229-9-137.
[21] Somta P, Seehalak W, Srinives P. Development, characterization and cross-species amplification of mungbean () genic microsatellite markers., 2009, 10: 1939– 1943.
[22] Wang L X, Elbaidouri M, Abernathy B, Chen H L, Wang S H, Lee S H, Jackson S A, Cheng X Z.Distribution and analysis of SSR in mung bean (L.) genome based on an SSR- enriched library., 2015, 35: 25, doi: 10.1007/s11032- 015-0259-8.
[23] Chen H L, Wang L X, Wang S H, Liu C Y, Blair M W, Cheng X Z. Transcriptome sequencing of mung bean (L.) genes and the identification of EST-SSR markers., 2015, 10: e0120273.
[24] Liu C Y, Fan B J, Cao Z M, Su Q Z, Wang Y, Zhang Z X, Wu J, Tian J. A deep sequencing analysis of transcriptomes and the development of EST-SSR markers in mungbean ()., 2016, 95: 527–535.
[25] 孙子奎, 陈永灿. 一种基于磁珠富集法高通量开发基因组SSR标记的方法. 中国专利, 2014, ZL201310222359.9. Sun Z K, Chen Y C. A method of developing genome SSR markers based on magnetic bead enrichment for NGS, China patent, 2014, ZL201310222359.9.
[26] Lindgreen S. AdapterRemoval: easy cleaning of next-generation sequencing reads., 2012, 5: 337, doi: 10.1186/ 1756-0500-5-337.
[27] Magoč T, Salzberg S L. FLASH: fast length adjustment of short reads to improve genome assemblies., 2011, 27: 2957–2963.
[28] Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth B C, Remm M, Rozen S G. Primer3: new capabilities and interfaces., 2012, 40: e115.
[29] Krawczak M, Nikolaus S, von Eberstein H, Croucher P J, El Mokhtari N E, Schreiber S. PopGen: population-based recruitment of patients and controls for the analysis of complex genotype-phenotype relationships., 2006, 9: 55–61.
[30] Liu K, Muse S V. PowerMarker: An integrated analysis environment for genetic marker analysis., 2005, 21: 2128–2129.
[31] 吴传书. 绿豆SSR标记的开发及高密度分子遗传连锁图谱的构建. 甘肃农业大学硕士学位论文, 甘肃兰州, 2014. Wu C S. Development of SSR Markers and Construction of a Genetic Linkage Map in Mungbean (L.). MS Thesis of Gansu Agricultural University, Lanzhou, Gansu, China, 2014 (in Chinese with English abstract).
[32] Gupta S K, Bansal R, Gopalakrishna T. Development and characterization of genic SSR markers for mungbean ((L.) Wilczek)., 2014, 195: 245–258.
[33] Yang T, Bao S Y, Ford R, Jia T J, Guan J P, He Y H, Sun X L, Jiang J Y, Hao J J, Zhang X Y, Zong X X. High-throughput novel microsatellite marker of faba bean via next generation sequencing., 2012, 13: 602, doi: 10.1186/1471-2164-13-602.
[34] Gao L F, Tang J F, Li H W, Jia J Z. Analysis of microsatellites in major crops assessed by computational and experimental approaches., 2003, 12: 245–261.
[35] Nicot N, Chiquet V, Gandon B, Amilhat L, Legeai F, Leroy P, Bernard M, Sourdille P. Study of simple sequence repeat (SSR) markers from wheat expressed sequence tags (ESTs)., 2004, 109: 800–805.
Development of SSR markers and genetic diversity analysis in mung bean
YE Wei-Jun**, CHEN Sheng-Nan**, YANG Yong, ZHANG Li-Ya, TIAN Dong-Feng, ZHANG Lei, and ZHOU Bin*
Crop Institute, Anhui Academy of Agricultural Sciences, Hefei 230031, Anhui, China
SSR markers play an important role in basic research and crop breeding due to their advantages of large number, high polymorphism and co-dominant inheritance. However, there are still few SSR markers available in mung bean. In this study, the magnetic bead enrichment method and sequencing technology were combined to identify the SSR loci of mung bean in high throughput, a total of 3,275,355 SSR loci were found, and 2742 markers were developed. A total of 157 markers were selected for validation by PCR method, 90 (57.33%) showed polymorphic among 10 mung bean accessions. Forty SSR markers with clear PCR products, high polymorphism and uniform distribution on chromosomes were selected to evaluate the genetic diversity among 90 mung bean accessions. The number of alleles per marker varied from two to eight, with an average of three. The effective number of alleles ranged from 1.31 to 4.21, with a mean value of 2.16. The Nei’s gene diversity was between 0.23 and 0.76, with an average of 0.51. Polymorphism information content was between 0.22 and 0.72, with a mean of 0.43. Cluster analysis distributed 90 materials into two clusters, including four groups. The germplasm of group II came from several areas, while those of groups I and III were mainly from North China and Shandong province, respectively. Most of the gerplasm from Hebei province were clustered in Group IV. These polymorphic SSR markers will be valuable for genetic diversity analysis, high-resolution genetic linkage maps construction, gene mapping and marker assisted selection in mung bean breeding.
mung bean; sequencing; SSR; primer design; genetic diversity
2018-11-21;
2019-01-19;
2019-03-16.
10.3724/SP.J.1006.2019.84155
周斌, E-mail:18756019871@139.com
**同等贡献(Contributed equally to this work)
叶卫军, E-mail: 963472965@163.com; 陈圣男, E-mail: chensn1226@163.com
本研究由安徽省农业科学院科研项目(18T0206), 国家现代农业产业技术体系建设专项(CARS-08-Z11)和国家重点研发计划项目(2016YFE0203800)资助。
This study was supported by the Research Program of Anhui Academy of Agricultural Sciences (18T0206), the China Agriculture Research System (CARS-08-Z11), and the National Key Research and Development Program (2016YFE0203800).
URL: http://kns.cnki.net/kcms/detail/11.1809.S.20190314.1427.008.html
猜你喜欢
今日农业(2022年13期)2022-09-15 01:18:00
世界科学技术-中医药现代化(2022年3期)2022-08-22 00:33:26
肝博士(2022年3期)2022-06-30 02:48:28
科学大众(2020年23期)2021-01-18 03:09:18
Journal of Sport and Health Science(2019年6期)2019-11-26 07:30:53
华人时刊(2018年15期)2018-11-18 16:31:42
启蒙(3-7岁)(2018年8期)2018-08-13 09:31:06
中国麻业科学(2018年6期)2018-04-09 11:22:12
西南农业学报(2016年5期)2016-05-17 05:42:21
广西林业科学(2016年3期)2016-03-16 05:43:21
