
Compartmentalized necroptosis activation in excitotoxicityinduced axonal degeneration: a novel mechanism implicated in neurodegenerative disease pathology
2019-07-17 02:13MacarenaS.Arrázola,FelipeA.Court
中国神经再生研究(英文版) 2019年8期
Excitotoxicity and neuronal cell death: Glutamate is the main excitatory neurotransmitter of the central nervous system and functionally involved in most brain activities, including brain development, synaptic plasticity, learning and memory. Excitatory synaptic transmission is primarily mediated by ligand-gated ion channels,including α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid(AMPA), N-methyl-D-aspartate (NMDA) and kainate receptors.Activation of glutamate receptors, particularly NMDA receptors,usually leads to calcium influx, which can act as a second messenger for several processes to mediate synaptic activity and brain function.Nevertheless, excessive release of glutamate neurotransmitter may produce intracellular calcium overload, leading to a cascade of events mediating cytoskeleton damage accompanied with reactive oxygen species (ROS) generation, mitochondrial dysfunction and ultimately neuronal cell death. These toxic effects of glutamate are known as excitotoxicity. Neuronal excitotoxicity has been linked to several acute and chronic brain diseases, such as stroke/ischemia, epilepsy and a range of neurodegenerative disorders, including Alzheimer's disease(AD), Huntington's disease, amyotrophic lateral sclerosis (ALS) and Parkinson's disease (PD), contributing to the neuronal lost in different brain regions. Unfortunately, treating nervous system disorders with general glutamate receptor blockers has been associated with undesirable side effects, becoming increasingly necessary to unravel downstream effectors in the excitotoxicity-dependent cell death pathway in order to develop novel therapeutic strategies.
As glutamate receptors are expressed in the neuronal soma,dendrites, axons and synaptic terminals, excitotoxicity could affect different neuronal compartments. In fact, glutamate excitotoxicity leads to neuronal apoptosis, as well as degeneration of axons and dendrites. Central myelinated axons express functional AMPA and GluR5 kainate receptors, and can directly respond to glutamate receptor agonists. These glutamate receptor-dependent signaling pathways promote an increase in intra-axonal Ca2+levels potentially contributing to axonal degeneration. Here, we discuss whether the compartmentalized destruction induced by excitotoxicity in neurons comprises different cell death mechanisms.
Unveiling the mechanism of axonal degeneration: The unique architecture of neurons, including in some cases meter-long axons,makes them extremely susceptible to damage. Axonal degeneration is a major contributor to neuronal dysfunction and a common and early feature of many neurological disorders, particularly those exhibiting excitotoxicity, as ALS, AD, PD and poly-glutamine disorders.
The degenerative process of mechanically damaged axons, known as Wallerian degeneration, has been extensively used as a model to unveil the cellular and molecular mechanisms of axonal degeneration. Studies in transgenic models displaying delayed Wallerian degeneration have demonstrated that axonal degeneration is a regulated self-destruction program. The expression of the Wlds protein is the most studied model of delayed axonal degeneration and emerging loss-of-function screens have also identified mutants that exhibit Wlds-like protection of injured axons, including dSarm/Sarm1 and Axundead (Osterloh et al., 2012; Neukomm et al., 2017). These models of delayed axonal degeneration have shown to protect in diverse neurodegenerative contexts, such as traumatic brain injury, chemotherapy-induced neuropathies and models of neurodegenerative diseases closely associated with neuronal death by excitotoxicity.
As axons degenerate early in several neuropathies and this degeneration can trigger neuronal death and disease progression, it is important to understand the mechanisms associated to this degenerative process. Despite axons can degenerate as a consequence of the activation of a ‘global' apoptotic program after somatic cell death(Geden and Deshmukh, 2016), it is currently known that ‘local' axonal degeneration or Wallerian degeneration is not apoptotic, which means that axonal destruction do not require caspases activation(Finn et al., 2000).
Like excitotoxicity-dependent neuronal damage, axonal degeneration is characterized by increase in ROS, axonal free calcium, and opening of the mitochondrial permeability transition pore (mPTP)(Barrientos et al., 2011). Interestingly, several events in the axonal degeneration program, including mitochondrial dysfunction, ROS production and calcium increase are common downstream effects of necroptosis.
Restricted necroptosis activation in axons upon excitotoxicity:Necroptosis was first described as a tumor necrosis factor alpha activated cell death signaling upon caspase-8 inhibitory conditions.Currently, it is known that necroptosis can be initiated by varieties of stimuli, including toll-like receptor ligands as lipopolysaccharide,endoplasmic reticulum stress, viral infection, increase in intracellular calcium and ROS. After activation, the kinases receptor-interacting serine/threonine protein kinase (RIPK) 1 and RIPK3 are phosphorylated, triggering phosphorylation and oligomerization of mixed lineage kinase domain-like pseudokinase (MLKL) which in turn leads to membrane permeabilization, cellular and organelle swelling,plasma membrane rupture and cellular contents release, inducing a pro-inflammatory response and cell death. Necroptosis participates in many nervous system pathologies that exhibit prominent axonal degeneration and excitotoxicity-induced neurodegeneration,such as multiple sclerosis, ALS, PD, AD, among others. The link between neuronal death induced by excitotoxicity and necroptosis was first proposed by Li et al. (2008). The authors showed in vitro that NMDA-induced excitotoxicity in cortical neurons die through a mechanism that involves necroptosis activation and calcium influx(Li et al., 2008). Nevertheless, beyond neuronal cell death, the mechanism by which excitotoxicity leads to early degeneration of axons remained unknown. Moreover, whether necroptosis is activated in a specific neuronal compartment, such as the axon, upon glutamate-induced excitotoxicity was unexplored until now. Our group recently demonstrated that excitotoxicity-induced axonal degeneration proceeds by a necroptotic mechanism, which activates mPTP,and calcium dysregulation in the axonal compartment without the requirement of caspase-8 inhibition. Pharmacological inhibition of RIPK1 with the canonical inhibitor Nec-1s, or by knocking-down RIPK3 or MLKL prevented key steps in the axonal degeneration cascade delaying axonal dismantling and thereby neuronal death.Surprisingly, experiments performed with hippocampal neurons seeded in microfluidic devices demonstrated that neuronal soma degenerates simultaneously by canonical apoptosis while compartmentalized necroptosis activation in axons mediates their degeneration, demonstrating differential death mechanisms in two cellular compartments under the same pro-degenerative stimulus (Hernández et al., 2018). These results are consistent with studies in other models of neuronal degeneration showing that soma and neurite degeneration are controlled by different degenerative mechanisms, in which neurite degeneration is regulated by a non-apoptotic process associated with mitochondrial dysfunction (Ikegami and Koike, 2003). Interestingly, we observed that inhibiting necroptotic factors prevents mitochondrial dysfunction induced by glutamate excitotoxicity in axons, including the loss of mitochondrial membrane potential and mPTP activation, suggesting that mitochondria are central effectors in axonal degeneration executed under excitotoxicity but also by diverse stimuli (Court and Coleman, 2012). Nevertheless, whether the necroptotic degeneration of axons after excitotoxicity crosstalk with the Wlds-dependent cascade remains to be elucidated.
Furthermore, it seems that even in the same cellular compartment the mechanism of excitotoxicity-induced neurodegeneration could have a differential regulation depending on the expression and distribution of glutamate receptors. In dendrites, it has been suggested that calcium flux through extra-synaptic NMDA receptors activates cell death pathways with features similar to necrotic cell death whereas synaptic receptor activation promotes cell survival and plasticity.Whether this regulation in axons occurs in a location dependent manner, for instance in axonal terminals compared with the axon shaft has not been explored. Furthermore, whether necroptosis regulators are differentially expressed along axons or dendrites remains to be defined.
Possible mediators of excitotoxicity-induced necroptosis activation in axons:Calcium is definitely a key player for axonal degeneration, since it is implicated in several stages of the degenerative process induced by excitotoxicity. Calcium regulates many physiological and relevant functions in the brain, but under certain conditions of deregulated ion concentrations, as occurs during excitotoxicity, its effect could be devastating for the neuron. Upon glutamate receptors over-activation, calcium influx exceeds the intracellular concentration that the endoplasmic reticulum and mitochondria are capable of buffering. Calcium overload in mitochondria induces the activation of the mPTP and uncontrolled calcium release, coupled with the loss of mitochondrial membrane potential, increase in ROS production, mitochondrial swelling and membrane disruption, leading to mitochondrial dysfunction, and rendering axons highly vulnerable to degenerate (Figure 1). On the other hand, it has been shown that calcium/calmodulin-dependent protein kinase II (CaMKII), a broadly expressed kinase in central nervous system neurons and the major regulator of neuronal calcium signaling, as well as glutamatergic excitotoxicity responses, induces RIPK1 phosphorylation in a model of calcium-induced viral infection in human neuroblastoma cells, leading to necroptosis activation and cell death (Nomura et al.,2014). Therefore, cytoplasmic calcium overload during excitotoxic stimuli might lead to necroptosis through RIPK1 phosphorylation after calcium-dependent CaMKII activation. Furthermore, it is well known that the first step in RIPK1 activation is the de-ubiquitylation of RIPK1 by protein cylindromatosis. As was discussed by Hernández et al. (2018), CaMKII-dependent protein cylindromatosis activation after NMDA excitotoxicity has been described in hippocampal neurons, suggesting calcium-dependent CaMKII activation as a crucial step in excitotoxicity-induced necroptosis activation to mediate axonal degeneration.
After RIPK1 activation, RIPK3 is phosphorylated to thereby phosphorylate MLKL, the downstream effector in the necroptotic signaling. MLKL is responsible for plasma membrane permeabilization,but also can translocate to other compartments, including mitochondria. Hernández et al. (2018) demonstrated that mPTP activation and loss of mitochondrial membrane potential occur downstream necroptosis, contributing to calcium homeostasis deregulation. Moreover,the necrosome complex can translocate to mitochondria and activate pyruvate dehydrogenase, increasing ROS generation, which in turn enhances necrosome formation and necroptosis activation (Yang et al., 2018). ROS therefore can function in a positive feedback loop that ensures effective induction of necroptosis (Figure 1).
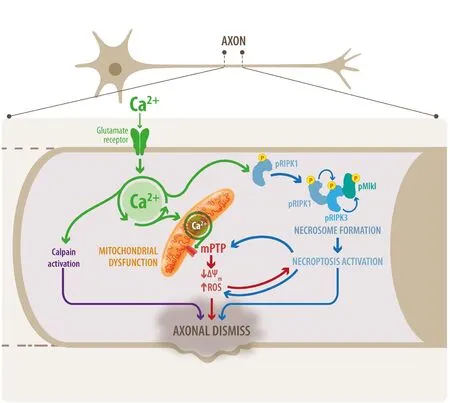
Figure 1 Necroptosis activators in excitotoxicity-induced axonal degeneration.Schematic representation of the factors that directly or indirectly activate necroptosis that can contribute to glutamate-induced axonal degeneration. Glutamate receptors over-activation induces excessive calcium influx in neurons. High calcium in the mitochondria together with the increased ROS production are ideal conditions to activate the mPTP, contributing to uncontrolled calcium release and mitochondrial membrane potential collapse, which affects axonal integrity contributing to axonal degeneration. Calcium can also directly activate calpains to promote axonal destruction or indirectly promote necroptosis activation through RIPK1 phosphorylation, which recruits their partners RIPK3 and MLKL to assemble the necrosome. Furthermore, necroptosis activation promotes mPTP activation and ROS production. ROS contributes with RIPK1 phosphorylation to enhance necrosome formation.Thus, ROS participates as a positive feedback loop that ensures effective induction of necroptosis to encourage axonal degeneration. ROS: Reactive oxygen specie; mPTP: mitochondrial permeability transition pore;RIPK: receptor-interacting serine/threonine protein kinase.
Prospects:Most therapeutic approaches against neuronal excitotoxicity are focused on glutamate receptor antagonists. Because glutamate is the major excitatory neurotransmitter, blocking the receptors affects neuronal function, with several side effects. A major goal of future research on excitotoxicity should be to understand how neurons can be protected without compromising their functionality. As axonal degeneration has been associated to several neurodegenerative conditions in their early phases, the participation of necroptotic-associated pathways unravel novel regulators of the axonal degenerative cascade, an important step to develop therapeutic strategies for excitotoxicity-associated nervous system disorders.
This work was funded by grants from Geroscience Center for Brain Health and Metabolism (FONDAP-15150012, to FC), Fondo Nacional de Desarrollo Científico y Tecnológico (FONDECYT, 1150766, to FC),Comisión Nacional de Investigación Científica y Tecnológica, Fondecyt de Postdoctorado Project (3180313, to MA)
Macarena S. Arrázola, Felipe A. Court*
Center for Integrative Biology, Faculty of Sciences, Universidad
Mayor de Chile, Santiago, Chile; FONDAP Center for Geroscience,Brain Health and Metabolism, Santiago, Chile (Arrázola MS, Court FA)Buck Institute for Research on Ageing, Novato, San Francisco, CA,USA (Court FA)
*Correspondence to: Felipe A. Court, PhD, felipe.court@umayor.cl.orcid: 0000-0002-9394-7601 (Felipe A. Court)
Received: December 29, 2018 Accepted: February 13, 2019
doi: 10.4103/1673-5374.253520
Copyright license agreement:The Copyright License Agreement has been signed by both authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- Improvement of ataxia in a patient with cerebellar infarction by recovery of injured cortico-ponto-cerebellar tract and dentato-rubro-thalamic tract: a diffusion tensor tractography study
- Tandem pore TWIK-related potassium channels and neuroprotection
- Dendritic shrinkage after injury: a cellular killer or a necessity for axonal regeneration?
- Regenerative biomarkers for Duchenne muscular dystrophy
- Exploring the efficacy of natural products in alleviating Alzheimer's disease
- Role of macrophages in peripheral nerve injury and repair