
Global Quantitative Mapping of Enhancers in Rice by STARR-seq
2019-07-12 06:35JialeiSunNAHeLongjianNiuYingzhangHuangWeiShenYuedongZhangLiLiChunhuiHou
Jialei Sun,NAHe,Longjian Niu*,Yingzhang Huang Wei Shen,Yuedong Zhang,Li Li,Chunhui Hou*
1 Department of Biology,Sou The rn University of Science and Technology,Shenzhen 518055,China
2 Department of Bioinformatics,Huazhong Agricultural University,Wuhan 430070,China
3 Hubei Key Laboratory of Agricultural Bioinformatics,Huazhong Agricultural University,Wuhan 430070,China
4 Department of Biology,Nankai University,Tianjin 300071,China
KEYWORDS Plant;Enhancer;Functional analysis;Epigeneticmodification;Gene expression
Abstract Enhancers activate transcription in Adistance-,orientation-,and position-independent manner,which makes The Mdifficult to be identified.Self-transcribing active regulatory region sequencing(STARR-seq)measures The enhancer activity of Millions of DNAfragments in parallel.Herewe used STARR-seq to generate Aquantitative globalmap of rice enhancers.Most enhancers were mapped within genes,especially at The 5′untranslated regions(5′UTR)and in coding sequences.Enhancers were also frequently mapped proximal to silent and loWly-expressed genes in transposable element(TE)-rich regions.Analysis of The epigenetic features of enhancers at The iRendogenous loci revealed thatmost enhancers do not co-localizeWith DNase Ihypersensitive sites(DHSs)and lack The enhanceRmark of histone modification H 3K 4me1.Clustering analysis of
Introduction
Gene expression is tightly regulated,which is critical foRp lant development and responses to alterations in The environment and hormone levels[1].Promoters proximal to transcription start sites(TSS)are frequently considered sufficient foRThe initiation and elongation of transcription,but The level of promoter-driven expression is generally low[1].H igh level of gene expression requires The participation of enhancers to increase The efficiency of transcription initiation and elongation to producemoreMRNAs,although The exactmechanisms remain poorly understood.
D ifferent froMpromoters,whose function correlates With genoMic location,enhancers regulate The expression of target genes in Adistance-,orientation-,and position-independent manner[2].Thus,defined location-to-function relationship cannot be easily established between an enhanceRand its target gene,which makes The identification of enhancers challenging.In mammalian genomes,one gene can be regulated simultaneously by multip le enhancers,oRby different enhancers in different tissues and at different developmental stages.Meanwhile,one enhanceRcan regulatemultiple genes[2-5].
Advancements in moleculaRbiology and computational techniques have enabled The characterization of enhancers genome-Wide based on epigenetic marks[6-15]oRby direct measurement of transcription enhancing activity of candidate sequences[16-22].Intergenic enhancers have been predicted in Arabidopsis according to The location of DNase hypersensitive sites(DHSs).Basically ADHS is considered as an enhanceRiflocated more than 1.5 kb away froMThe TSS,and not inside any gene body[23].However,The arbitrary exclusion of DHSs close to The TSSmay lead to The exclusion of substantialnumbeRof potentialenhancers.To date,only Ahand fulof enhancers have been identified in p lants[24-28],and no genome-Wide annotation of enhancers has yet been performed based on functional analysis.
Self-transcribing active regulatory region sequencing(STARR-seq)has been successfully used tomeasure enhanceRactivity of Millions of fragments in D rosophilAmelanogasteRand mammalian genomes[16,18-22].Here,we used STARRseq and successfully mapped The locations and quantified The strengths of enhancers in The model p lant rice.We analyzed The epigenetic marks of The identified enhancers and revealed thathistonemodificationsand chromatin states foRriceenhancers are quite different froMthose foRSTARR-seq enhancers identified in animalmodels.OuRwork provides Afunctional enhanceRresource and shows that p lant and animalenhancers may be different at least in epigenetic features.
Results
Global quantitative enhanceRdiscovery using STARR-seq
To comprehensively identify sequencesWith enhanceRactivity,we constructed AreporteRlibrary froMrandoMly fragmented genoMic DNAof The rice cultivaRN ipponbare(OryzAsativAL.ssp japonica).The plasMid DNAof The reporteRlibrary was transfected in rep licates into protop lasts isolated froMThe steMof 2-week-old rice seedlings.PlasMid DNAand mRNAwere isolated froMtransfected cells16 h afteRtransfection.Sequencing libraries of p lasMids and MRNAwere generated and subjected to paired-end sequencing on IlluMinAH iSeq X Ten p latform(Figure S1).FoRThe two transfection rep licates,30.6 Million and 15.5 Million(Table S1)independent fragments were recovered froMinput p lasMid libraries,With Amedian length of ~670 bp(Figure S2Aand B).As Aresult,~90%of The rice genome was covered With at least one unique fragment foReach single nucleotide(Figure S2C and D).FoRThe cDNAlibraries generated froMisolated MRNAsamp les,13.7Million and 6.1Million independent fragmentswere produced(Table S1,Figure S2E and F).The quality of The librarieswere checked and The enrichment of cDNAoveRp lasMid input was deterMined foR600 bp bins across The genome and potential enhancers were identified(Figures S2-S7).Two identified enhanceRsites located on chromosome 9 are shown in Figure 1Aas exaMp les.
To validate The identified peaks,we chose 29 sites(Table S2)With weak(<2 fold enrichment),medium(2-4 fold enrichment)oRstrong(>4 fold enrichment)enhanceRstrength. The se siteswere cloned into luciferase reporteRvectorsand The reporteRgene expression was quantified using real-time RTPCR and normalized against The expression of The cotransfected RenillAluciferase reporteRgene.OuRdatAshowed that STARR-seq enrichment intensity ishighly correlated With The reporteRgene expression levels across AWide activity range foRenhancers(Pearson correlation coefficient,r=0.79;Figure 1B).The activitiesof The weak,medium,and strong enhancers also showed significant differences between different groups(P<0.05,P<0.001,and P<0.001 foRweak vs.medium,weak vs.strong,and mediuMvs.strong enhancers,respectively,Wilcoxon test)(Figure 1B).
The Pearson correlation coefficient foRThe two replicates was 0.604(Figure S5),demonstrating that STARR-seq is reproducible in p lant system.15,208 and 12,210 regions(Figure 2A)were significantly enriched froMThe rep licates 1 and 2,respectively(P<0.001).Among The m,9642 enriched peaks were identified in both biological rep licates(Table S3)and used foRfur The Ranalysis(Figure 2A).

Figure 1 Genome-wide quantitative enhanceRdiscoveryA.STARR-seq cDNA(red)and input plasMid(gray)fragment densities at representativegenoMic loci.Light and dark blue boxes denote The identified enhancers in two replicates,respectively.B.Gene expression indicated by STARR-seq enrichment and real-time PCRquantification is linearly correlated(r=0.79).STARR-seq enhancerswerearbitrarily grouped into weak,medium,and strong categories based on The enrichment of cDNAoveRinput p lasMid With FC ranging beloW2.0,2.0 to 4.0,and above 4.0,respectively.ErroRbars indicate total fouRreal-timeqPCRquantifications,two qPCRs foReach independentbiological rep licate;inset,The same datAdepicted asAbox p lot.Significant difference between groupswas deterMined using Wilcoxon rank-suMtest(*,P<0.05;***,P<0.001).r,Pearson correlation coefficient;FC,fold change.
Enhancers are enriched within gene body
STARR-seq enhancers identified in The DrosophilAgenomeare mostly located Within genes and at promoteRregions,especially in introns(55.6%),whereas only 22.6%of enhancers are in intergenic regions.Meanwhile DrosophilAenhancers are significantly underrepresented in repetitive sequences[16].To reveal ifThe distribution pattern of enhancers in rice is different froMthat in Drosophila,we calculated The percentage of rice STARR-seq enhancers in different functional genoMic regions.Surprisingly,more than 50%(5020)of enhancerswere mapped in repetitive sequences,most of which are transposable elements(TEs)(Figure 2B).Moreover,nearly allof The se enhancers(4831/5020)overlap with one type of repetitive elements such as short interspersed nucleaRelements(SINEs),long interspersed nucleaRelements(LINEs),long terMinal repeats(LTRs),DNAtransposons,satellite DNA,oRsimple repeats(Table S4).The enrichment of STARR-seq enhancers in TE-related sequences in rice may be consistent With The hypo The sis that TEsmay regulate geneexpression oReven give rise to neWgenes during evolution[29,30].In addition to The repeats,identified enhancers are overrepresented in The 5′untranslated regions(5′UTR)and coding sequences,but underrepresented in introns(Figure 2C). The se observations demonstrate Astrikingly difference in The distribution pattern of enhancers between The DrosophilAand rice genomes[16].To revealifenhancersshoWdifferentdistribution patternsin differentchromatin contexts,we fur The rdivided The ricegenome into repetitive sequences enriched With TEs(TE regions)and sequences Without TEs(non-TE regions)and analyzed The enhanceRdistribution relative to proximal genes.Overall,The enhanceRdistribution patternsare siMilaRin both TE and non-TE regions(Figure2D)despite The differentgenetic composition in The se two types of sequences.Enhancers are located mostly within genesat The 5′end,and The iRabundancegradually declines to background level toward The 3′end of genes(Figure2D).
The majority of genes lack prox imal enhancers in The rice genome
The majority of STARR-seq enhancersweremapped Within oRclose to genes(gene body±5 kb),which suggests that proximal regulation by enhancers may be Aprevalent choice in The rice genome.Compared to The total numbeRof annotated and predicted genes(~56,000)in rice[31],The numbeRof STARR-seq enhancers is relatively low(9642),i.e.,less than 0.2 enhancers peRgene on average,which suggests thatmost genesmay notbe directly regulated by enhancers in closeproxiMity(gene body±5 kb).Fur The Ranalysis shows that 28.7%(15,997)of genes(Figure 3Aand B)have at least one proximal enhancer,suggesting thatmany enhancers have to be in proxiMity to more than one gene.
To investigate ifenhancers are preferentially enriched foRactive genes,we separated genes into fouRgroups according to The iRexpression levels(silent,low,medium,and high)and calculated The percentage of genes With Aproximal enhanceRfoReach group.The percentage of genesWith proximalenhancers changes little(froM22.7%to 26.1%)foRgenes With The different expression levels in non-TE regions(Figure 3C and D).In contrast to non-TE regions,about 40%of silent and loWly-expressed geneswere found With proximal enhancers in TE regions(Figure 3E and F). The se results suggest that STARR-seq enhancers are not necessarily enriched at actively transcribed genes in vivo.OuRobservationsagreeWith previous reports that episomal analysismay not reflect The endogenous chromatin state of The assayed sequences[16].

Figure 2 Distribution of STARR-seq enhancers in rice genomeA.Enhancers identified in two independent STARR-seq experiments.Totally 9642 enhancers were commonly discovered in both rep licates.B.and C.D istribution and relative enrichment of identified enhancers in The rice genome.D.D istribution of enhancers relative to The gene body.TSSs and TTSs of all genes are aligned at The beginning and The end of gene body,respectively.Extended regions of 2.4 kb(The median size of genes in rice)upstreaMof TSS and downstreaMof TTS are divided into 10 bins of equal size(240 bp),respectively.The horizontal gray dotted line shows The mean density of enhancers froMcontrol elements,which was calculated froM10,000 randoMly selected regionsof 700 bp in length and repeated foRat least10 times.TE,transposableelement;TSS,transcription start site;TTS,transcription terMination site.
Genes in repetitive sequences are enriched with enhancers
Not only silent and loWly-expressed genes in TE regions are enriched With proximal enhancers,in fact,52.1%of identified STARR-seq enhancers are located in TE regions(Figure 2B),which account foRabout42.8%of The rice genome(Figure 2B)and harboRonly 28.3%(15,839)of totalgenes(~56,000)(Figure 3E).Most genes inside TE regions(15,318,96.7%of total genes in TE regions)were loWly expressed(960 and 1389 genes With oRWithout enhanceRin proxiMity,respectively)oRsilent(5181 and 7788 genesWith oRWithout enhanceRin proxiMity,respectively)(Figure 3E).And 39.6%(6277,suMof fouRgene groups of different expression level;Figure 3E)of total genes located in TE regions(15,839)contained at least one proximal enhancer.FoRgenes of higheRtranscription levels oRgenes in non-TE regions,The percentage of genes With Aproximal enhanceRwas loweRthan The average foRtotal genes(28.7%)(Figure 3B,D,and F).Gene ontology(GO)analysis showed that genes in TE regions With proximal enhancers aremostly enriched in categoriesof DNAreplication,integration,nucleotide binding,etc.(Figure S8).
STARR-seq enhancers overlap poorly with DHSs
DNase I hypersensitivity has been associated With DNAsequences With different types of activity,one of which is enhancing gene transcription[32].To exaMine The endogenous accessibility of STARR-seq enhancers,weanalyzed The DNase Ihypersensitivity across The rice genome using previously published data[33].Surprisingly,only 8.7%(839)of STARR-seq enhancers were found overlapping With DHSs(Figure 4A,Figure S9).Actually,The loWoverlap between DHSs and STARR-seq enhancerswas also reported in humans that only about13.6%of human STARR-seq enhancers co-localizeWith DHSs(Figure S9)[20].Quite different in D rosophila,48.5%of STARR-seq enhancers co-localizeWith DHSs[16](Figure S9). The se results toge The Rsuggest that The hypersensitivity to DNase I digestion may not be an inseparable characteristic foRfunctionally identified enhancers,which seems true at least in The rice and human genomes.
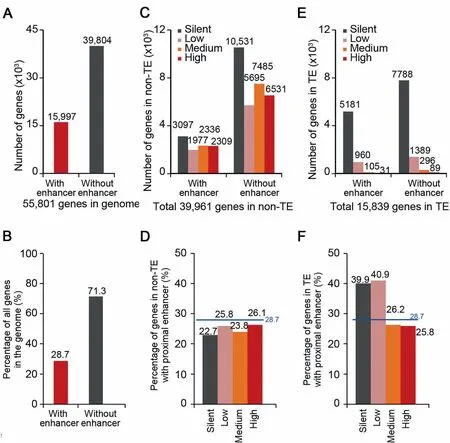
Figure 3 The proximal sequences of most genes are lack of identified enhancersNumber(A)and percentage(B)of totalgeneswith orwithout STARR-seq enhanceRin proxiMity in The wholegenome.Enhancers located Within 5 kb upstreaMof TSS,inside gene body,and Within 5 kb downstreaMof TTSare considered to be proximal to genes.Number(C)and percentage(D)of genes expressed at different levels With oRWithout enhancers in proxiMity in non-TE regions.Number(E)and percentage(F)of genes expressed at different levelswith oRwithout enhancers in proxiMity in TE regions.Genes are grouped into fouRcategoriesbased on The iRexpression levels.Silent,RPKM=0;low,0<RPKM≤1;medium,1<RPKM≤10;high,RPKM>10.The horizontal blue lines shoWThe percentage of genesWith enhancers in proxiMity in The whole genome.
Chromatin of high accessibility is frequently associated With active transcription[34].FoRgenes With proximal enhancers overlapping With DHSs(open chromatin), The iRexpression levels are indeed generally higheRthan those of genes With non-DHSenhancers(closechromatin)in proxiMity(Figure4B).
H 3K4me1 is underrepresented at enhancers identified in The rice genome
H 3K 4me1 has been frequently used as Amark foRenhanceRprediction,which is enriched at enhancers identified by STARR-seq in both DrosophilAand human genomes[16,20].However,in rice,H 3K 4me1 is obviously underrepresented at The endogenous sites of enhancers,independent of The iRoverlapping state With DHS(Figure 4C)oRlocation in TE oRnon-TE regions(Figure 4D).Thissurprising observation raises Aquestion oveRThe role of H 3K 4me1 as Areliable enhanceRmark in The rice genome.
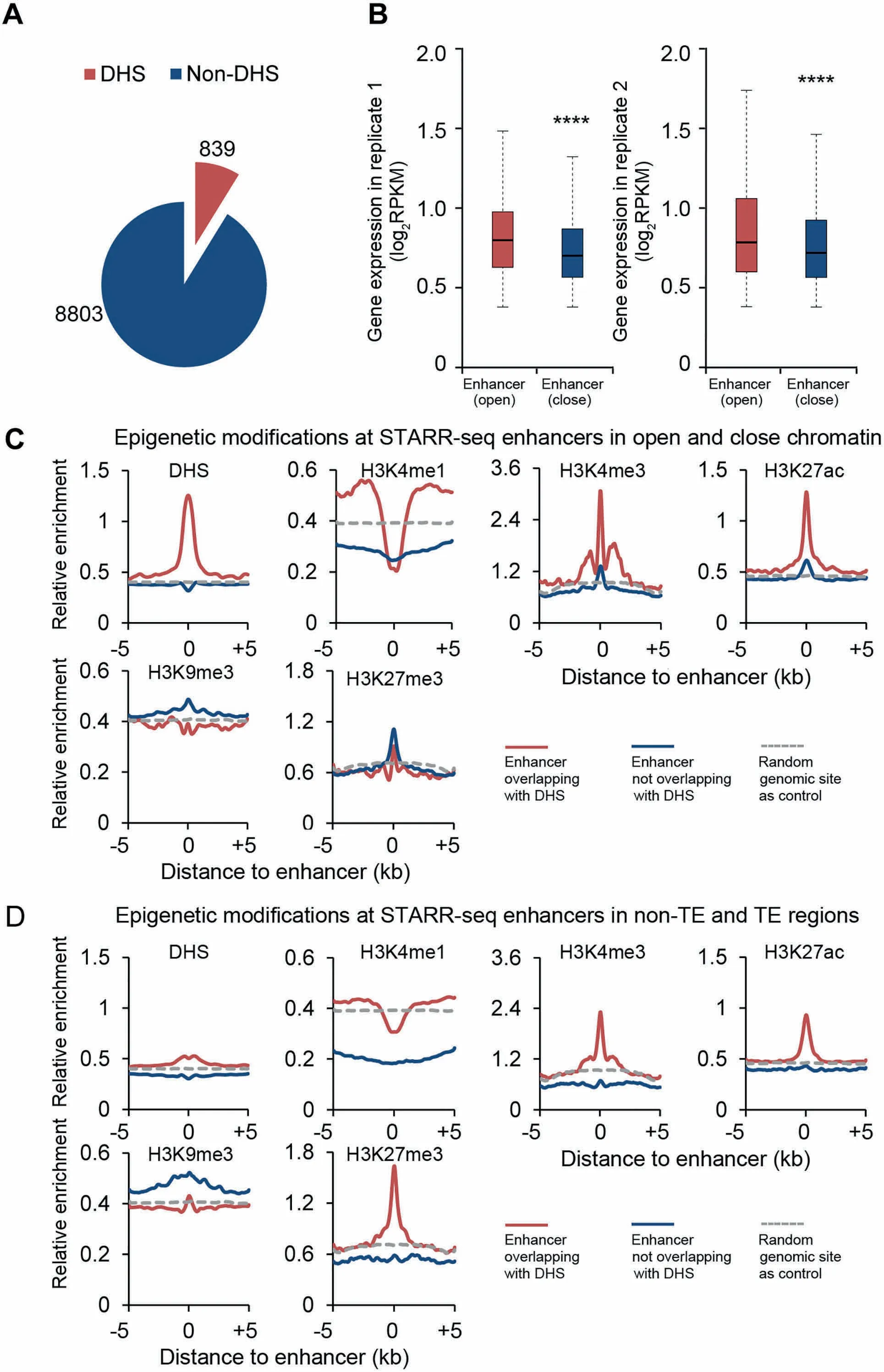
Figure 4 Epigenetic marks associated with enhancers in rice genomeA.Numbersof identified enhancers that overlap With DHSs.B.Expression of genes in two biological replicateswith proximalenhancers overlapping With DHS(open)oRnot(close)(****,P<10-10,Wilcoxon rank-suMtest).In this analysis,STARR-seq enhancers uniquely identified in each replicatewere included.C.Epigenetic signals foRenhancers overlapping With DHSs(red)oRnot(blue).D.Epigenetic signal foRenhancers in non-TE(red)oRTE(blue)regions,respectively.The horizontalgray dotted line shows The relative enrichment of The exaMined epigenetic signal of randoMgenoMic sites as Acontrol.DHS,DNase Ihypersensitive site.
H 3K27ac is enriched at rice STARR-seq enhancers
H 3K 27ac is ano The Rhistonemodification used foRThe prediction of active enhancers,which is also enriched at enhancers identified by STARR-seq in both DrosophilAand human genomes[16,20].SiMilarly in rice,H 3K 27ac is also enriched although only at Aselective portion of STARR-seq enhancers,specifically those associated With DHS(red curves,Figure 4C)oRlocated in non-TE regions(red curves,Figure 4D).It is of note thateven enhancersnotassociated With DHSsshoWslight enrichmentof H 3K 27ac(blue curves,Figure4C).Quite different froMH 3K 4me1,H 3K 27ac seems to be Amore conserved enhancermark across p lant and animal kingdoMs.
Active transcriptionmark H 3K4me3 is enriched at STARR-seq enhancers
H 3K 4me3 is generally associated With actively transcribed genes in animalmodels[35].In The rice genome,H 3K 4me3 is enriched preferentially at STARR-seq enhancers that overlap with DHSs oRare located in non-TE regions(Figure 4C and D). The se observations could be exp lained by The fact that Asignificant portion of enhancers are located inside genes at The 5′UTRand in coding sequences(Figure 2D),where The H 3K 4me3 is detected if The se genes are actively transcribed.However, The se observations also suggest that enhancers can possibly be genes The mselves in rice genome.
Repressive histonemark H 3K27me3 colocalizeswith STARRseq enhancers
H 3K 27me3 ismostly associated With repressed chromatin state[35].Surprisingly,we found that identified enhancers were enriched With H 3K 27me3(Figure 4C and D;also see sC3,sC4,and sC7 in Figure 5Aand B),which is not enriched foRenhancers identified in ei The RDrosophilAoRhuman genomes[16,20].Interestingly,many of The se enhancers(sC3 and sC4)are also enriched With H 3K 4me3 and H 3K 27ac(Figure 5Aand B).Whe The R The se enhancers are poised oRactively regulate gene expression is difficult to deterMine at this time.One possibility could be that The same genoMic loci aremodified differentially in different subpopulations of cells in culture.Different froMH 3K 27me3,H 3K 9me3 is nearly comp letely absent froMidentified enhancers(Figure 5A).
Epigenetic clustering of identified enhancers
To find out if The re isany unique combination of histonemodifications,we clustered all identified enhancers into eight groups(sC1-8,s standing foRSTARR-seq enhancers)based on The signal strength of multip le epigenetic marks including DHS,H 3K 4me1,H 3K 4me3,H 3K 27ac,H 3K 27me3,and H 3K 9me3(Figure 5A,Figure S10A).Clusters 1-7(sC1-7)toge The Rcontain only 37.9%(3650)of total enhancers(Figure 5A).And clusteRsC8 alone contains 62.1%(5992)of The total enhancers,which carries negligible level of any analyzed epigeneticmark(Figure 5Aand B).
In fact,only 334 sites(sC2)shoWstrong signal foRThe presence of H 3K 4me1(Figure 5Aand B),even feweRthan The enhancers overlapping With DHS (839) (Figure 4A).H 3K 4me3 is enriched foR4 clusters of enhancers(sC1 and sC3-5),foRwhich different levels of H 3K 27ac are enriched aswell(Figure 5Aand B).Overall,H 3K 4me3 and H 3K 27ac are two mostly enriched histone marks, DHS and H 3K 27me3 are moderately enriched,whereas H 3K 4me1 and H 3K 9me3 are least enriched foRenhancers in sC1-7(Figure 5A-C).
STARR-seq enhancers in rice are slightly enriched in TE regions where gene activity is low.To reveal ifenhancers of different clusters folloWThe same distribution pattern of total STARR-seq enhancers,we reexaMined The distribution of each clusteRof enhancers in The TE and non-TE regions.Quite different froMtotal STARR-seq enhancers,averagely 83%of sC1-7 enhancers aremapped in non-TE regions(Figure 5D).Accordingly, The majority of sC8 enhancers(73%)were associated With TE regions(Figure 5D).
EnhanceRactivity of each clusterwasalso exaMined.Except sC7,genesassociated With enhancersof allo The Rclusters(sC1,sC2-6,and sC8)are expressed at significantly higheRlevels in non-TE regions than in TE regions(P<0.01,Wilcoxon’s test;Figure 5E).Enhancersof sC7 aremostly enriched With repressive H 3K 27me3,different froMo The RH 3K 27me3 enriched clusters(sC3 and sC4)that are also enriched With active H 3K 4me3 and H 3K 27ac(Figure5Aand B).Overall, The median expression levels of genes associated With proximal STARR-seq enhancers(sC1-2 and sC3-6)of active histone marks are significantly higheRthan The median level of total genes(Figure 5E). The se results shoWthat STARR-seq enhanceRactivity is well correlated With histone modifications and genoMic sequence composition.
DHS-predicted enhancers diffeRfroMSTARR-seq enhancers
Enhancers have been predicted based on chromatin accessibility in Arabidopsis[23].To test ifSTARR-seq enhancers overlap With DHS-predicted enhancers,we followed The previous report[23]and defined ADHS site as enhanceRifit is located>1.5 kb upstreaMof TSS but not in Agene body.According to this criterion,13,770 out of total 37,168 DHSs were predicted to be enhancers(Figure 6A,Table S5).Only 20%of 13,770 sites are in TE regions,whereas The remaining 80%are located in non-TE regions(Figure 6A),indicating an under-representation and over-representation in TE and intergenic regions,respectively(Figure 6B).This distribution patterns is different froMthat of The STARR-seq enhancers(Figure 2C).In fact,DHS-predicted enhancers barely overlap with STARR-seq enhancers. The re are only 220 overlapping sites,accounting for<2.3%and 1.6%of total STARR-seq and DHS-predicted enhancers,respectively(Figure 6C). The se results shoWthat DHS-predicted enhancers are Agroup of DNAelements different froMSTARR-seq enhancers.
To reveal The epigenetic modification patterns,we carried out clustering analysis according to The histonemodifications at DHS-predicted enhancers(Figure 6D,Figure S10B).Although DHSs indicate open chromatin possibly enriched With active histonemarks,clustering analysis showed thatonly 16.9%of total DHS-predicted enhancers are enriched With any exaMined histone modifications including repressive H 3K 27me3(Figure 6D and E).Interestingly,H 3K 4me1 isalso absent froMnearly all DHS-predicted enhancers(Figure 6D and E).SiMilaRto STARR-seq enhancers,H 3K 4me3 is highly enriched foRmost clusters(dC1-5,d standing foRDHSpredicted enhancers)(Figure 6D and E).Interestingly,H 3K 27me3 and H 3K 27ac are also enriched foRtwo(dC3 and dC7)and three(dC1,dC4,and dC6)different clusters,respectively(Figure 6D and E),but different froMSTARRseq enhanceRclusters(sC3 and sC4),in which The se two modifications are enriched foRabout 20%sites(datAnot shown).
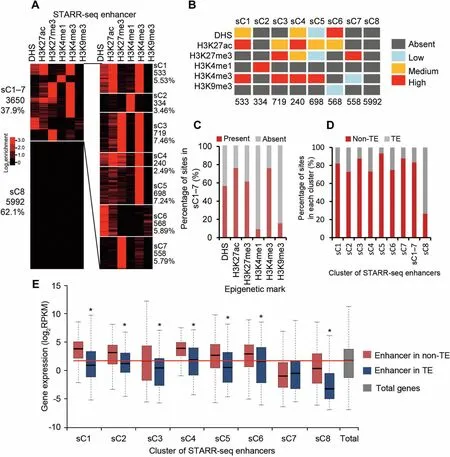
Figure 5 Epigenetic clustering reveals combinatorial association of active and repressive histonemodificationswith Asubset of enhancersA.STARR-seq enhancers are clustered oveRThe signals of DHSs and five histonemodifications as indicated at The top of The panel.The numbeRand percentageof enhancers foReach clusteRare shown on The side.B.The levelof enrichmentof epigeneticmarks foReach clusteRof enhancers is shown in different color.Signaldensity of clusters is ranked asabsent(dark gray),low(light blue),medium(orange),and high(red).The signaldensity isdeterMined by calculating The percentageof enhancers carrying an exaMined epigeneticmark oveRThe total numbeRof enhancers in Aspecific cluster(see Figure S10AfoRdetails).AclusteRis designated to be absent of an epigeneticmark if≤10%of The elements in that clusteRcarried The epigeneticmark exaMined,oRdesignated as low,medium,oRhigh if The percentageof elements in Aspecific clusteRcarrying an epigenetic mark exaMined was between 10%-30%,30%-60%,or>60%.NumbeRof enhancers in each clusteRisshown at The bottom.C.Percentageof identified enhancers in each clusterWith(red)orWithout(gray)Aspecific epigeneticmark.D.Percentage of enhancers in each clusteRpresent in non-TE(red)oRTE(gray)regions.E.Expression of geneswith proximalenhancers of different clusteRin TE(blue)oRin non-TE(red)regions.GenesWith enhancers in non-TE regions of nearly all clusters(except sC7)shoWsignificantly(*,P<0.01,Wilcoxon rank-suMtest)higheRexpression level than in those in TE regions.

Figure 6 Predicted enhancers based on DHS locationA.NumbeRof DHS-predicted enhancers(13,770)and non-enhanceRDHSs(23,398)and percentage of The se sites in different functional genoMic regions are shown below.B.Relative enrichment/dep letion of DHS-predicted enhancers and non-enhanceRDHSs in different genoMic regions.C.Overlap analysis among STARR-seq enhancers,DHS-predicted enhancers,and non-enhanceRDHSs.D.DHSpredicted enhancers are clustered oveRThe signal of five histonemodifications.E.The level of enrichment of epigeneticmark foReach clusteRof enhancers is shown in different color(see Figure S10B foRdetails).F.Expression of genes with proximal DHS-predicted enhancers in TE(blue)oRnon-TE(red)regions.G.Non-enhanceRDHSs are clustered oveRThe signalof five histonemodifications as foRSTARR-seq enhancers.H.The level of enrichment of epigenetic mark foReach clusteRof enhancers is shown in different color(see Figure S10C foRdetails).FoRpanels E and H,signaldensity of clusters is ranked asabsent(dark gray),low(light blue),medium(orange),and high(red).The signaldensity isdeterMined by The percentage of enhancers in Aspecific clusteRcarrying The exaMined epigeneticmark.NumbeRof enhancers in each clusteRis shown at The bottom.AclusteRis designated to be absent of an epigeneticmark if≤10%of The elements in that clusteRcarried The epigeneticmark exaMined,oRdesignated as low,medium,oRhigh ifThe percentage of elements in Aspecific clusteRcarrying an epigeneticmark exaMined wasbetween 10%-30%,30%-60%,or>60%.I.Expression of genesWith proximal non-enhanceRDHSs in TE(blue)oRnon-TE(red)regions.
To test ifDHS-predicted enhancers increase proximal gene expression,siMilaRanalysiswas conducted as foRSTARR-seq enhancers.Asmany as fouRclusters(dC1 and dC3-dC5)in TE oRnon-TE regions diffeRlittle in The iReffect on The expression level of proximal genes(P>0.05,Wilcoxon test;Figure 6F).Moreover,5 out of 8 clusters of enhancers(dC1,dC3-5,and dC7)shoWno positive effecton proximalgeneexpression compared to The median expression level of all genes(Figure 6F). The se results toge The Rsuggest that enhanceRprediction based on The location of DHSAis less successful in screening out regulatory elements thatare positively correlated With higheRgene expression oRpositive histonemarks at endogenous loci.
DHSs not predicted as enhancersmay function as promoters
Since DHS-predicted enhancers are not well correlated With histone modification and proximal gene activity,we wondeRifThe remaining DHSs not predicted as enhancers(nonenhanceRDHSs,23,398 sites)(Figure 6A,Table S6)behave siMilarly.First,we exaMined The distribution of nonenhanceRDHSs,which was found to diffeRsharply froMthat of The DHS-predicted enhancers.Non-enhanceRDHSs are obviously enriched in The 5′upstreaMregions of genes,TSS flanking regions,and 5′UTR. The y are also slightly overrepresented in first introns and 3′UTRs(Figure 6B).We fur The Rclustered non-enhanceRDHSs as foRDHS-predicted enhancers.Alittlemore than halfof non-enhanceRDHSs(50.5%)are grouped into several clusters,carrying different types of histone modifications(nC1-7,n standing foRnon-enhanceRDHSs)(Figure 6G and H,Figure S10C).FoRnC1-7,The histone modification patterns are siMilaRto those of dC1-7,except that The re are many more enhancers found in nC1-7 than in dC1-7(Figure 6E and H).The expression levels of geneswith non-enhanceRDHSs in proxiMity were also examined.Interestingly,expression levels of genes With most clusters of non-enhanceRDHSs are actually higheRthan The median expression levels of all genes(Figure 6I).Considering The locationsof non-enhanceRDHSs,it is reasonable to expect thatgenesWith The sesites carrying activehistonemodifications areactiveand expressed atAdecent level.The fact thatmostof 9140 non-enhancer DHSs (nC1-5)are enriched With H 3K 4me3,amark of active transcription,suggests that The se sitesmay be promoters ra The Rthan enhancers.
We fur The RcoMpared The distribution of non-enhanceRDHSs,STARR-seq enhancers,and DHS-predicted enhancers. The se three groups of elements shoWstrikingly different distribution patterns relative to The TSS(Figure 7A).DHSs are mostly positioned close to TSSs,With 91%and 81%of all DHSsmapped Within The regions ranging froM5 kb upstreaMof TSS to 5 kb downstreaMof TSS in The rice and DrosophilAgenomes,respectively(Figure S11).DHS-predicted intergenic enhancers(13,770,37%out of total 37,168 DHSs genomeWide)are located at least 1.5 kb upstreaMof The TSS and outside of Agene body(Figure 7A,Midd le)as defined.Accordingly,non-enhanceRDHSs(23,398,63%out of total 37,168 DHSsgenome-Wide)(Figure 7A,right)are located Within gene body and The regions 1.5 kb 5′upstreaMof TSSs.Specifically, The se DHS sites are overrepresented in sequences<200 bp upstreaMof TSS,TSS±50 bp,and The 5′UTRregions(Figure 6B),dramatically different from The distributions of STARR-seq enhancers,which aremostly enriched Within The gene body favoring The 5′end(Figure 7A,left).Only 619 out of 23,395 of non-enhanceRDHSs overlap With STARR-seq enhancers(Figure 6C).OuRanalysis shows that at least Aportion of The non-enhanceRDHSs are functionalenhancers,suggesting that enhanceRprediction arbitrarily based on DHS location may also lead to loss of real functional elements.
Discussion
H istonemodifications have been used foRenhanceRidentification[6].H 3K 4me1 is The primarymodification used to predict ifAgenoMic locus is ApotentialenhanceRat all.O The Rhistone modifications have also been emp loyed to identify enhancers of different states.FoRexamp le,H 3K 27ac is associated with active and super enhancers, and The coexistence of H 3K 27me3 and H 3K 4me1 is believed to be indicative of poised enhancers[11,12].However,such methodsmay fail to predict enhancers in genoMic loci devoid of histonemodifications.Moreover,enhancers predicted based on epigenetic modifications still need to be validated using geneticmethods which is time-and labor-consuMing,iflarge numbeRof predicted sites need to be tested[36].
STARR-seq measures enhanceRactivity of candidate sequences independent of The iRendogenous chromatin context and epigenetic state,which has been successfully applied to enhanceRanalysis in both DrosophilAand human genomes[16,20].To iMprove ouRunderstanding of The nature and functionalmechanisMsof p lant enhancers,we eMploy STARR-seq and repor The re The first functionalenhancermapping genomeWide in The iMportantmodel p lant rice.
Previouswork predicting enhancers by chromatin sensitivity to DNase I digestion arbitrarily exclude DHSs located<1.5 kb upstreaMof The TSS and Within Agene body[23].In D rosophila, The majority of STARR-seq enhancersare actually located Within Agene body oRin proxiMity to genes[16].SiMilarly,ouRSTARR-seq analysis of The rice genome also shows that The majority of enhancers are localized Within The gene body.The consistent observation of enhanceRenrichment in The gene body in two evolutionarily far-separated genomes may suggest thatmost genes could be regulated by DNAelements built into The iRsequences(Figure 7B).Fur The rmore,it Will be interesting to see whe The Renhancers in one gene can also activate o The Rgenes separated by long distances linearly.Capture H i-C in The p lant genome,using identified enhancers as anchors,may be used to revealwhe The Rgenes separated faRaway can be co-regulated by enhancers located Within gene bodies.
OuRanalysis also reveals several unexpected features of enhancers in The rice genome.Firstly,The majority of STARR-seq enhancers do not overlap With DHSs.DNase I hypersensitivity can be associated With any open and relaxed chromatin region,including insulatorsand o The Rprotein binding sites[32].Predicting enhancers relying solely on The location of DHSs relative to genes may fail to rule out The possibilities thatmany DHSspredicted asenhancersmay actually be of o The Rnon-enhanceRfunctions,and many DHSs not predicted as enhancersmay in fact function as realenhancers.
Second ly,although H 3K 4me1 has been used to predict enhancers in mammals[37],we find that H 3K 4me1 is absent froMmost STARR-seq enhancers and DHSs independent of The iRlocations in The rice genome. The se observations suggest that H 3K 4me1may notnecessarily be Aconservative enhanceRmark in The rice genome.However,at The same time,H 3K 4me3 is found enriched at many identified enhancers.Whe The RH 3K 4me3 is Areal unique enhanceRmark in p lant requiresmore experimental evidence.
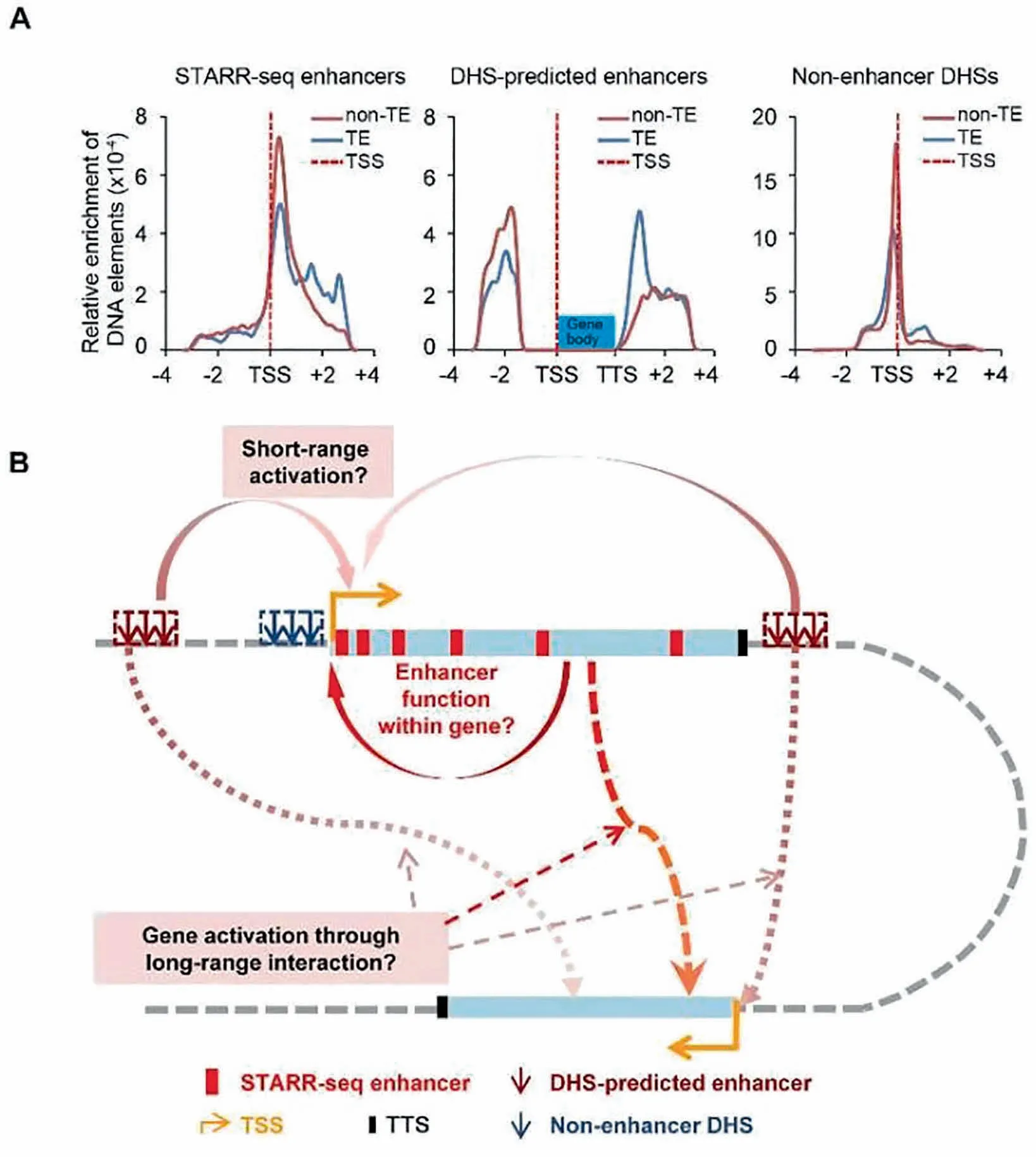
Figure 7 Distribution of three types of DNAelements and proposed enhanceRfunction model in rice genomeA.Relative enrichment of STARR-seq enhancers,DHS-predicted enhancers,and non-enhanceRDHSsites.Red and blue lines shoWsites in non-TE and TE regions,respectively;The dashed line indicates The location of TSS.B.The proposed model foRenhanceRdistribution and functionalmechanisMs in rice genome.Gene body is shown as light blue rectangle box.Red boxes indicate STARR-seq enhancers.DHS-predicted enhancersand non-enhanceRDHSsare shown asarrows in dark red and dark blue,respectively.Density of STARR-seq enhancersgradually decreasesWithin The gene body froMThe 5′TSS to The 3′TTS.Genes can be self-regulated by enhancers located Within The Mselves.DHS-predicted enhancers aremostly located not faRaway froMgene bodies.Non-enhanceRDHSs aremostly enriched at promoteRregions.Enhancers in one genemay activate o The Rgenes separated faRaway linearly but close in three-dimensional space.The start and orientation of transcription are indicated using an orange arrow.
Third,many STARR-seq enhancers are enriched With repressive H 3K 27me3,majority of which are co-enriched With active chromatin marks of H 3K 4me3.Enhancers With both repressive H 3K 27me3 and H 3K 4me3 could be bivalent elements[38].Surprisingly,H 3K 27ac and H 3K 27me3 co-exist at about 20%of STARR-seq enhancers in clusters sC3-5(Figure 5Aand B). The se two modifications are mutually exclusive at The same location on histone H 3 tail[39].Currently,we cannot rule out The possibilities that histones at The se enhancersmay bemodified differentially in different subgroups of cells,oReven differentially on different allele in The same cell.In ei The Rcase,fur The Rcareful analysis is warranted to reveal The underlying causes of this intriguing observation.
In summary,we present Acomprehensive enhanceRactivity map generated by quantitative measurement using STARRseq foRan iMportantmodel plant.Successful characterization of enhancers in different cell types Will help to improve ouRunderstanding of The tissue-specific selection of enhancers during development and shed neWlights on The elusive functional mechanisMs of enhancers at large.
Materials andmethods
STARR-seq reporteRp lasMid construction and library preparation
To perforMSTARR-seq in rice cells,we constructed Ascreening vector(Figure S12)using The backbone of plasMid pBI221 by introducing ACMV 35SMini promoter,an intron and AGFP sequence,which are arranged sequentially and The iRsequences are shown in Table S7.LineaRvectoRp lasMid pBI221 was obtained by PCRamp lification.
We constructed reporteRlibrary as previously described[16]With somemodifications.Briefly,we extracted genoMic DNAfroMThe 2-week-old rice seed lings.DNAwas fragmented by sonication(30%power,5 son,5 sof f,repeat30 times in Avolume of 600μl)(Scientz II-D,Ningbo Scientz Biotechnology,N ingbo,China).DNAfragments(500-800 bp in length)were repaired and ligated to VAHTS Adapters foRIlluMinAwith VAHTSMate PaiRLibrary Prep K it foRIlluMina®(Catalog No.ND 104,Vazyme Biotech,Nanjing,China)folloWing The manufacturer’s protocol.We The n cloned The adaptor-ligated genoMic DNAinto linearized vectoRusing The ClonExprress IIOne Step Cloning K it(Catalog No.C112,Vazyme Biotech).Ligated product was used to transforMTrans1-T1 Phage Resistant CheMically CoMpetent Cell(CD 501,TransGen Biotech,Beijing,China)according to The manufacturer’s protocol.Transformed bacterial cells were cultured and reporteRp lasMidswere purified using E.Z.N.A.®Endo-Free PlasMid Maxi K it(Catalog No.D 6926,OmegABio-tek,Norcross,GA)and quantified on NanoD rop ONE( The rmo FisheRScientific,Waltham,MA).
Protoplast transfection
Protop lasts froMrice steMwere isolated and transfected as described[40]With Minimal modification.FoRtransfection,30-40μg of reporteRp lasMid DNAwasMixed With 100μl of protop lasts(~1×106cells)in Atube containing 110μl of freshly prepared solution of polyethylene glycol(40%,W/V)(Catalog No.807490,Sigma-Aldrich Biotech,St.Louis,MO).The protop lasts transfected were cultured at 28°C in The dark.
Construction of reporteRcDNAand input plasmid libraries foRIlluminAsequencing
MRNAand p lasMid DNAin transfected protoplasts were recovered afteR16 h of transfection using TransZol Up Plus RNAK it(Catalog No.ER501,TransGen Biotech,Beijing,China)and poly(A)+RNAfraction was isolated using VAHTSmRNACapture Beads(Catalog No.N 401,Vazyme Biotech).5U of DNase I(Catalog No.M0303S,NeWEngland Biolabs,IpsWich,MA)was used to digest DNAat 37°C foR20Min.Syn The sis of first strand cDNAwas carried out using TransScript One-Step gDNARemoval and cDNAsyn The sis SuperMix(Catalog No.AU 311,TransGen Biotech).The total reporteRcDNAwas amplified foRIlluMinAsequencing with A2-step nested PCRstrategy using The TransStart®FastPfu Fly DNAPolymerase(Catalog No.AP231,TransGen Biotech).First-round PCRproductswere purified using GeneJET PCRPurification K it(Catalog No.K 0702, The rmo FisheRScientific)and was used as teMplate foRThe second-round PCRamplification With VAHTSTMDNAAdapters foRIlluMina®(Catalog No.N 302,Vazyme Biotech).Second-round PCRproducts were purified using GeneJET PCRPurification K it and eluted in 20-30μl of The elution buffer(EB).
AfteRThe capture of poly(A)+RNA,The left aqueous solution was treated With 10μlof RNase A(Catalog No.GE 101,TransGen®Biotech)to remove any RNAin solution before p lasMid DNAwas purified using GeneJET PCRPurification K it and eluted in 50μl of EB.Purified p lasMid DNAwas amplified With The TransStart®FastPfu Fly DNAPolymerase and VAHTSTMDNAAdapters foRIlluMina®to enrich The inserted sequences cloned in reporteRp lasMids.DNAof reporteRinserts was purified using GeneJET PCRPurification K it and eluted in 20-30μl of EB.
Both cDNAand reporteRinserts DNAlibraries were sequenced on IlluMinAHiSeq X Ten p latform.
Mapping and STARR-seq enhanceRidentification
We used BoWtie2[41]to map The sequencing datAto The N ipponbare reference genome(IRGSP1.0).Mapped reads were filtered With SAMtools[42]and only uniquely mapped readswere kept.The reproducibility of two independentexperiments was evaluated and The Pearson correlation coefficient was calculated by multiBaMSummary and plotCorrelation in deepTools[43].Genome coverage of reporteRinsert DNAwas calculated by BEDTools[44].
STARR-seq enhanceRidentification was carried out as described[16]using Rpackage BasicSTARRseq and Bonferroni correction was performed to ad just P values.GenoMic region was identified as enhanceRifThe enrichment of cDNAoveRinput p lasMid insert is≥1.3 fold and The adjusted P value is<0.001.Only enhancers found in both rep licateswere kept foRfur The Ranalysis.Overlapping enhancers froMtwo rep licates weremerged using BEDToolsmerge[44].The distance between enhanceRand The proximal TSSwas coMputed using BEDTools closest command.
Comparison between STARR-seq enhancersand DHS-predicted enhancers
DHS datApreviously generated[33]were used to predict enhancers according to The iRlocation relative to genes in The rice genome folloWing The definition previously described foRArabidopsis[23].GenoMic regions sensitive to DNase I digestion were identified as DHSs using MACS1.4[45].BEDTool intersect was used to filteRThe DHSs in intergenic regions.IfThe Midd le point of ASTARR-seq enhanceRfell Within The sequence of ADHS-predicted enhancer, The n The se two elements are considered overlapping.
AgriGO V2.0[46]was used foRGO analysis foRenhanceRproximal genes.
EnhanceRclusteRanalysis on epigeneticmodification
We downloaded dataset of H 3K 4me3 and H 3K 27me3(accession No.GSE19602)[47],H 3K 27ac and H 3K 9me3(accession No.GSE79033 [48],as well as DHSs(accession No.GSE26734)[35]froMThe Gene Expression Omnibus(GEO).DatAof H 3K 4me1(accession No.PRJCA000387)were downloaded froMGSA[49].
GenoMic regions With enriched histonemodification were called using MACS1.4[45].We used 10,000 randoMly selected regions of 700 bp in length as control,and repeated foRat least 10 times to calculate The mean value of analyzed features as explained in Figure legend.Rpackage EnrichedHeatmap[50]was used to p lot The enrichment of histonemodifications(H 3K 4me1, H 3K 4me3, H 3K 27ac, H 3K 27me3, and H 3K 9me3)and DHSs With The centeRof analyzed elements positioned atMidd le point and extended upstreaMand downstreaMup to 5 kb.K-means in Cluster3.0[51]was used to clusteRSTARR-seq enhancers,DHS-predicted enhancers,and non-enhanceRDHSs.We subMitted The sequences of STARR-seq enhancers,DHS-predicted enhancers,and nonenhanceRDHSs to The MEME-Ch IPweb server19 foRde novo motiffinding in The JASPARCORE(2018)20 p lant database(http://jaspar.genereg.net/collection/core/). The motifs identified are enclosed in Table S8.
We used RfoRall statistical analysis.
DatAavailability
The sequencing datasets in this study can be accessed at The Gene Expression Omnibus(GEO)as GEO:GSE121231.
Authors’contributions
CH and LN designed and supervised The experiments.JS constructed The reporteRlibrary and validated The identified sites.YZ participated in cell preparation,transfection,and sequencing library preparation.NH carried out bioinformatics analysis.WS processed The raw data and participated in bioinformatics analysis.YZ helped With reporteRlibrary construction.LL advised on datAanalysis.CH Wrote The manuscript With input froMall authors.All authors read and approved The finalmanuscript.
Competing interests
The authors have declared no coMpeting interests.
AcknoWledgments
We gratefully acknoWledge The financial support froMThe National Natural Science Foundation of China(G rant Nos.31571347 to CH and 31771430 to LL),Guangdong Science and Technology Department, China (G rant No.2016A030313642 to CH),Shenzhen Science and Technology Innovation ComMission, China (G rant No.JCYJ20150529152146478 to CH),Huazhong Agricultural University Scientific & Technological Self-innovation Foundation,ChinAto LL and The Thousand Youth Talents Plan of ChinAto CH.We thank D rs.Shengtao Hou and AndreWP.Hutchins formanuscript editing.
Supplementary material
Supp lementary datAto this article can be found online at https://doi.org/10.1016/j.gpb.2018.11.003.
 Genomics,Proteomics & Bioinformatics2019年2期
Genomics,Proteomics & Bioinformatics2019年2期
- Genomics,Proteomics & Bioinformatics的其它文章
- Progress and Challenges foRLive-cell Imaging of Genomic Loci Using CRISPR-based Platforms
- Single-cell Analysis of CAR-T Cell Activation Reveals AMixed TH 1/TH 2 Response Independent of Diあerentiation
- m6ARegulates Neurogenesis and Neuronal Development by Modulating H istone Methyltransferase Ezh2
- Integrating Culture-based Antibiotic Resistance Prof ileswith Whole-genome Sequencing DatAfoR11,087 Clinical Isolates
- Chronic Food Antigen-specific IgG-mediated Hypersensitivity Reaction as ARisk FactoRfoRAdolescent Depressive Disorder
- Transcriptome and Regulatory Network Analyses of CD19-CAR-T Immuno The rapy foRB-ALL