
微波提取-高压液相色谱-原子荧光光谱联用(HPLC-AFS)法分析稻米样品中砷形态
2019-07-06 07:39魏洪敏甄长伟炼晓璐李银贺
中国无机分析化学 2019年3期
魏洪敏 甄长伟 炼晓璐 李银贺 柴 刚
(北京海光仪器有限公司,北京100015)
前言
元素的毒性与元素的存在形态息息相关,就砷元素而言,无机砷的毒性要大于有机砷[1-2]。因此,在分析砷元素时,其形态分析是十分必要的,比用砷的总量来评价更具科学性。近年来联用技术已经成为元素形态分析的主要手段。高效液相色谱-原子荧光光谱联用法可以对砷元素各形态进行有效分离,然后再逐一检测,从而实现样品的在线分离与测定。目前该方法已用于土壤[3]、大米[4]、水产动植物[5]、地下水[6]等样品中砷元素的形态分析。
食品安全国家标准GB 5009.11—2014《食品中总砷和无机砷的测定》,应用的是液相色谱-原子荧光光谱联用法[7]。本文围绕该标准,使用液相色谱-原子荧光光谱联用仪,对样品前处理方法、流动相、氢化反应条件等进行了优化。并在最优条件下对稻米及稻米标准物质进行分析测定,也对方法的精密度、加标回收率、检出限等指标进行了验证,测定结果令人满意。
1 实验部分
1.1 仪器与试剂
LC-AFS 6500液相-原子荧光联用仪(北京海光仪器有限公司)、微波消解仪(上海新仪微波化学科技有限公司)、真空抽滤装置(天津奥特赛斯仪器有限公司)、BGZ-76电热鼓风干燥箱(上海博讯实业有限公司)、高性能空心阴极灯(北京有色金属研究总院)、pH计、离心机等。
磷酸二氢铵、磷酸二氢钾、十二水合磷酸氢二钠、氢氧化钠、盐酸(优级纯,北京化工厂)、硝酸(优级纯)、硼氢化钾,除另有说明,所用试剂均为分析纯,且购买于国药集团化学试剂有限公司。
砷形态溶液标准物质购买于中国计量科学研究院:亚砷酸根[GBW08667,(75.7±1.2) μg/g],二甲基砷酸[GBW08666,(52.9±1.8) μg/g],甲基砷酸[GBW08669,(25.1±0.8) μg/g],砷酸根[GBW08667,(17.5±0.4) μg/g],含量均以As计。
1.2 样品前处理
称取约1.0 g样品于微波消解罐中,加20 mL硝酸溶液(0.15 mol/L),用微波消解仪90 ℃下恒温提取50 min。提取完毕后冷却至室温,于8 000 r/min离心15 min,取上清液,过0.45 μm滤膜,使用液相色谱-原子荧光光谱联用仪进行测定。
1.3 分析条件
砷形态的测定条件如表1所示。

表1 砷形态测定条件
2 结果与讨论
本文对影响测量的条件进行了优化,包括样品前处理、流动相选择、氢化物发生条件。
2.1 实验条件的优化
2.1.1 样品前处理
形态分析的前处理一定要保证被测组分不能发生形态转变。选取了某糙米样品(含As(III)和DMA两种目标组分),使用稀硝酸进行提取,研究了热浸提取和微波提取两种方法。
热浸提取:称取约1.0 g样品于50 mL离心管中,加入20 mL硝酸溶液(0.15 mol/L),放置过夜。于90 ℃恒温箱中热浸提2.5 h,每30 min振摇1 min。提取完毕,取出冷却至室温,于8 000 r/min离心15 min,取上清液,过0.45 μm滤膜,使用液相色谱-原子荧光光谱联用仪进行测定。
微波提取:按样品处理方法处理样品,分别提取了20、30、40、50、60 min,结果表明,50 min时,目标组分已基本提取完全。
两种方法经过对比分析发现,微波提取与热浸提取的测定结果相当,鉴于微波提取大大缩短了样品前处理的时间,最终选用了微波提取,且提取时间为50 min。
2.1.2 流动相
2.1.2.1 流动相的选择
不同的元素形态具有不同的理化性质和毒性,不同形态的砷化合物能够被离子交换色谱分离的主要原因是:在适宜的pH值下,砷的化合物能够在流动相中电离,以不同离子形态存在于水溶液中。通常,对流动相有如下要求:1)对被分析物具有合适的溶解度,并提供离子交换所必须的缓冲溶液;2)对被测离子有不同的洗脱能力,能从分离柱中依次取代这些离子;3)要有合适的离子强度,以控制各目标分析物的保留时间。离子交换色谱一般都以各种盐类的缓冲溶液作流动相,通过调节缓冲溶液类型、pH值、离子强度,以及通过加少量有机溶剂等增加对被分析物的分离选择性,使待测样品达到良好的分离效果[8]。
使用Hamilton阴离子交换柱,以15 mmol/L磷酸二氢铵(pH=6)为流动相,分离测定砷元素的4个形态(As(III)、MMA、DMA和As(V)),分析时间约为15 min。为提高效率缩短分析时间,对流动相进行了调整。参照文献[9],选取了KH2PO4-Na2HPO4缓冲体系,并摸索了其对砷形态分离的影响。最终确定的流动相组成为KH2PO4(45 mmol/L)-Na2HPO4(5 mmol/L),使用该流动相砷形态4个组分能够在7 min内达到基线分离,分析时间远远低于国家标准“GB5009.17—2014”中给出参考的12 min。
2.1.2.2 流动相的配制
NH4H2PO4(15 mmol/L,pH=6):称取1.7g NH4H2PO4,溶于1 000 mL水中,用氨水调节pH值为6.0。
KH2PO4(45 mmol/L)-Na2HPO4(5 mmol/L):称取3.026 g KH2PO4和0.895 4 g Na2HPO4·12H2O,溶于500 mL水中,混匀,溶液的pH值为5.91,无需调节。
2.1.2.3 流动相的优化
按表2的比例配制流动相(pH值实测值与表中吻合),分别使用上述流动相对砷形态标准溶液进行洗脱,LC-AFS的其余测定条件见表1。测定结果见图1,从上至下对应流动相的pH值分别为:5.29、5.59、5.91、6.24、6.47、6.64、6.81、9.18,从图中可知:1)随着流动相pH值的增大,As(III)保留时间基本保持不变,As(V)的保留时间逐渐缩短;2)pH值为5.91时,各组分分离效果最好;3)pH值较低时,As(III)和DMA分离不好;4)pH值较高时,DMA和MMA分离不好,甚至两色谱峰完全重叠。

表2 KH2PO4-Na2HPO4缓冲溶液
为获得更好的分离效果,在KH2PO4和Na2HPO4比例为9∶1的条件下,对流动相进行稀释以降低离子强度。实验了总浓度为50 mmol/L和25 mmol/L的流动相,其pH值均为5.91,并与表格中的1/15 mol/L(总浓度为66.7 mmol/L)的流动相进行对比分析,各组分的分离效果见图2。从图2可知,随着流动相中盐浓度的降低,各组分的分离效果变好,但某些组分的保留时间延长且峰展宽严重。综合考虑选择了50 mmol/L[KH2PO4(45 mmol/L)-Na2HPO4(5 mmol/L)]的缓冲盐作为流动相。
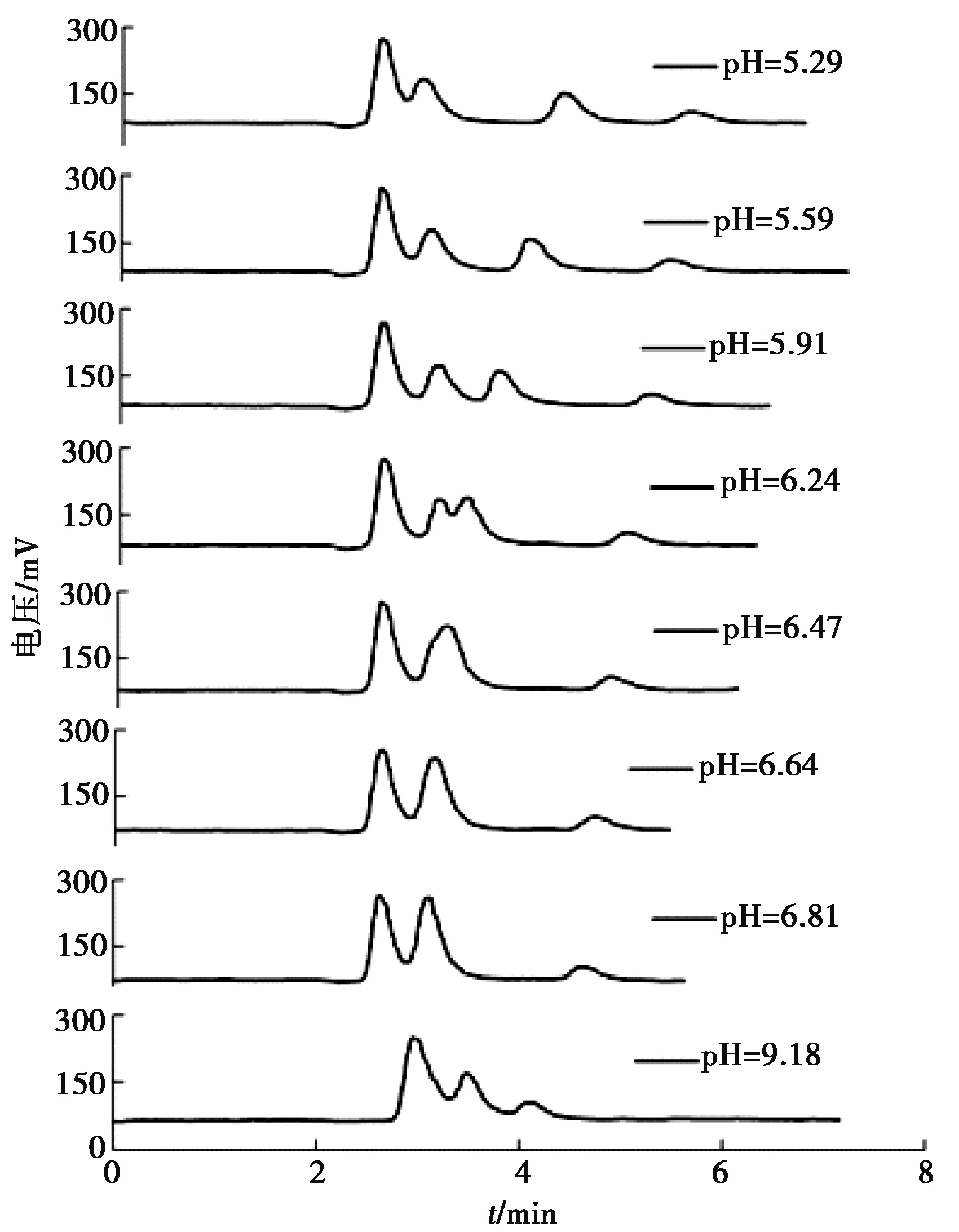
图1 流动相对砷形态分离效果的影响Figure 1 Effect of mobile phase on the separation efficiency of arsenic speciation
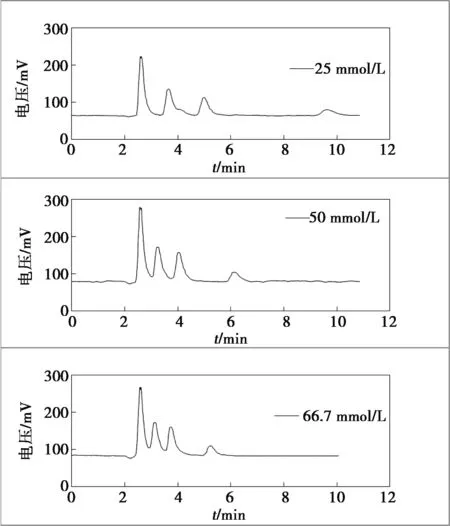
图2 离子强度对分离效果的影响Figure 2 Effect of ionic strength on the separation efficiency of arsenic speciation.
2.1.3 氢化反应条件
2.1.3.1 载流
以盐酸为载流,研究了其浓度对各形态峰面积的影响。从图3可知,当载流浓度为2%时,峰面积较小,可能是由于酸浓度低时,反应速率低导致响应值低;载流浓度为5%时,峰面积最大。载流浓度大于10%时,各组分的峰面积逐渐降低,可能是由于反应过程中产生的大量氢气稀释了原子化器中砷元素的浓度。但当载流为10%时谱图的基线更平稳。最终,选择载流浓度为10%。
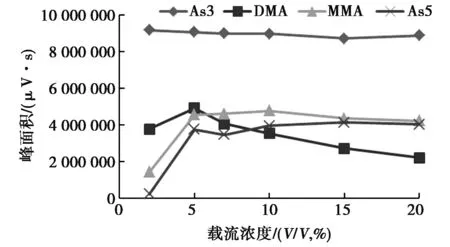
图3 载流浓度对峰面积的影响Figure 3 Effect of concentration of carrier currenton the peak area.
2.1.3.2 还原剂
还原剂的作用是在一定的酸度下将砷元素各形态还原为氢化物。研究了KBH4溶液浓度对砷形态峰面积的影响。从图4可知,当KBH4浓度为20 g/L和40 g/L时,各组分的峰面积相对较高。考虑到40 g/L时“氢化反应”较剧烈,基线波动较大,最终选择了20 g/L的KBH4作为还原剂。当KBH4浓度为10 g/L时,响应值都偏低,这可能是由于氢化反应效率较低;当KBH4浓度较高时,可能是由于反应过程中产生的大量氢气稀释了原子化器中砷原子的浓度,使得峰面积偏低。

图4 还原剂浓度对峰面积的影响Figure 4 Effect of concentration of reducing agent on the peak area.
2.1.3.3 载气
实验研究了载气流速对各组分峰面积的影响。从图5可知,随着载气流速的增加,各组分的峰面积都逐渐降低,当载气流速为200 mL/min时各组分的峰面积最大;在实验中发现流速为200 mL/min时,会引起较大的基线噪声和漂移。随着流速的继续增高,原子蒸汽被稀释,峰面积值降低。最终,选取300 mL/min为载气流速。

图5 载气流速对峰面积的影响Figure 5 Effect of speed of carrier gas on the peak area.
2.1.3.4 屏蔽气
屏蔽气能保持火焰形状稳定。研究了屏蔽气流速对各组分峰面积的影响。从图6图可知,屏蔽气流速为900 mL/min时,4个组分的峰面积均最大。因此,选用900 mL/min为屏蔽气流速。

图6 屏蔽气流速对峰面积的影响Figure 6 Effect of speed of shieldinggas on the peak area.
2.2 方法学考察
2.2.1 线性范围与检出限
考察方法的线性范围和检出限(以3倍的基线噪声比计),结果见表3。配制一系列砷形态的标准样品:浓度为2、4、6、8和10 ng/mL,绘制标准曲线,结果表明,各组分在测定范围内线性关系良好,相关系数均大于0.998 8。检出限分别为0.29、0.47、0.62和1.16 ng/mL。
2.2.2 精密度实验
对同一浓度的砷形态标准溶液连续进样7次,将峰面积测定结果列于表4中,以峰面积RSD衡量定量重现性。从表4可知,峰面积RSD均低于3.1%,表明方法重现性较好。

表3 线性范围与检出限

表4 方法精密度实验
2.3 实际样品分析
按照实验方法分析3个市售大米样品以及2个糙米样品,将分析结果汇总于表5中。从表5数据可知,在所测定的样品中,As(III)都有检出、DMA和As(V)部分检出、MMA均为未检出,且所有样品中总无机砷的含量均未超过“GB 2762—2012”中规定的限定值[10](该标准中稻谷、糙米、大米中总无机砷的限量为0.2 mg/kg)。

表5 样品测定结果
另外,对稻米标准物质进行测定,该标准物质证书中总无机砷为(0.298±0.008) mg/kg,二甲基砷酸为(0.018 6±0.000 8) mg/kg。实际测定总无机砷的含量为0.297 mg/kg,DMA的含量为0.019 mg/kg,可见结果都在不确定度范围内,说明该方法对无机砷的测定结果准确。
同时,对市售大米样品和糙米样品进行加标回收实验,添加水平为2 ng/mL和4 ng/mL,并将加标回收结果列于表6和表7中。结果表明,4个组分在两类样品中的回收率都较好,回收率为85.1%~113%。
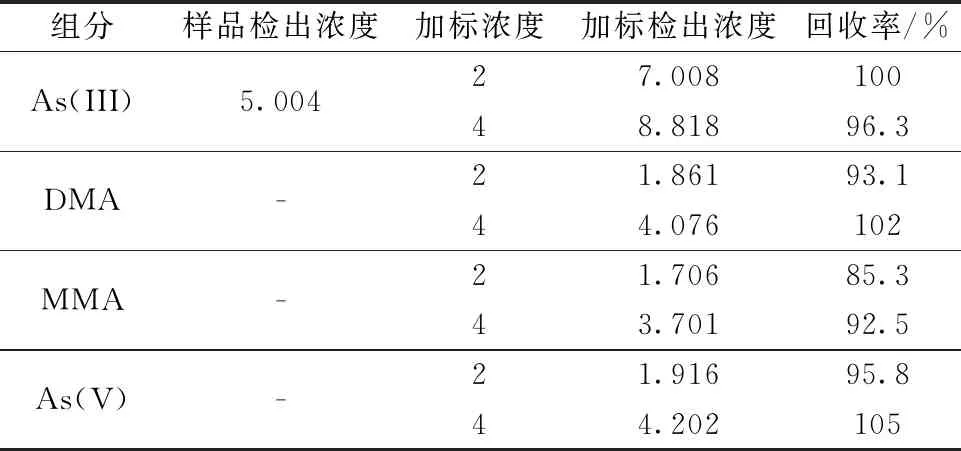
表6 市售大米1加标回收测定结果
2.4 方法比较
建立的微波提取法与GB 5009.11—2014的对比结果见表8。从该表8可知,本实验所选择的微波提取法,前处理时间短、分析速度快,使用的载流和还原剂浓度也较低。

表7 糙米样品1加标回收测定结果

表8 方法比较
3 结论
对GB 5009.11—2014中的样品前处理方法及砷形态测定的影响因素进行了优化研究。建立了更加高效环保的微波提取法,在最优条件下对稻米样品进行了测定,通过方法学考察和加标回收等实验,验证了该方法的有效性。结果表明该方法的线性范围、检出限、精密度和加标回收率等指标均良好,说明改进的方法完全可以用于稻米中砷形态的测定。
猜你喜欢
初中生学习指导·提升版(2022年4期)2022-05-11
中学生数理化·八年级物理人教版(2022年4期)2022-04-26
煤气与热力(2021年12期)2022-01-19
环境卫生工程(2021年3期)2021-07-21
大众科学(2020年7期)2020-10-26
当代化工(2019年3期)2019-12-12
小天使·六年级语数英综合(2018年1期)2018-10-08
电子制作(2018年10期)2018-08-04
红领巾·探索(2018年12期)2018-01-26
电子制作(2017年13期)2017-12-15
