
RP-HPLC测定注射用硫普罗宁的有关物质
2019-07-03 04:38:26许晓明徐莉莉
中国药科大学学报 2019年3期
安 冏,李 堪,许晓明,高 准,胡 超,李 博,3,狄 斌,3*,徐莉莉,3**
(1中国药科大学药物分析系,南京 210009;2江苏复旦复华药业有限公司,海门 318099;3中国药科大学药物质量与安全预警教育部重点实验室,南京210009)
硫普罗宁[N-(2-巯基丙酰基)-甘氨酸](TP)是一种含巯基的化合物,属罕见病用药。其化学结构见图1-a。目前,TP已广泛应用于多发性肝病[1]、胱氨酸尿症[2]、类风湿性关节炎[3]和白内障[4]。它还用于预防肾脏损害[5],作为重金属中毒的解毒剂[6]以及防辐射剂[7]。然而,TP存在许多不良反应,如过敏反应[8]、肾病综合征[2]、肝毒性[9]和贫血伴随血液异常[10]。值得注意的是,该药物的一些不良反应是由TP的有关物质(RS)引起的,例如TP引起的过敏性休克,是由生产过程中的游离硫醇或杂质引起的[11]。自1988年在美国上市以来,TP的质量标准至今尚未建立。因此,迫切需要一种有效的方法来检测注射用硫普罗宁中的有关物质,便于控制TP质量,减少不良反应。
本研究建立了一种新的、简便的、灵敏的高效液相色谱法分析注射用硫普罗宁中的有关物质,并对其进行了验证。该方法与现有报道方法(包括HPLC[12]、LC-MS[13]、GC-MS[14]、荧光法[15]、化学发光法[16]、分光光度法[17]、伏安法[18]和流动注射分析[6]等)相比,具有明显优势。而且,在分析过程中揭示了巯基化合物和甘氨酸类似物在反相色谱上的保留行为以及流动相pH对待测物色谱行为的影响。根据国际协调会议(ICH)准则对该方法进行了验证,并对实际样品进行测定。本研究可为国内外药典注射用硫普罗宁质量标准的制定提供重要参考。
1 材 料
1.1 药品与试剂
TP对照品(纯度≥99.0%,中国药品生物制品检定所);注射用硫普罗宁(江苏复旦复华药业有限公司,河南信义药业有限公司);TP原料(山东洛新制药集团有限公司)。杂质对照品(其化学结构如图1所示):2-氯丙酰甘氨酸(RS 1,纯度≥97.0%,武汉丰泰威源科技有限公司);2-巯基丙酸(RS 5,纯度≥97.0%,美国西格玛-阿尔德里奇公司);硫普罗宁二硫化物(RS 7,纯度≥98.0%,加拿大多伦多研究化学品公司)。乙腈(HPLC级,美国Tedia公司);其余试剂均为市售分析纯。去离子水和蒸馏水由美国PALL纯水仪制备。

Figure1 Chemical structures of (a) tiopronin (TP),(b) 2-chloropropionylglycine (RS 1),(c) 2-mercaptopropionic acid (RS 5) and (d1-d3) TP disulfide (RS 7)
1.2 仪 器
LC-20 AT高效液相色谱仪,SPD-M20A光电二极管阵列检测器(PDA)(日本岛津公司);6520系列四极杆-飞行时间质谱仪(Q-TOF,美国Agilent公司);Thermo Scientific Syncronis C18柱(250 mm×4.6 mm,5 μm,美国Thermo公司);Waters Symmetry C18柱(250 mm×4.6 mm,5 μm,美国Waters公司)。
2 方法与结果
2.1 色谱-质谱条件
Waters Symmetry C18分析柱(250 mm×4.6 mm,5 μm),乙腈-0.1%甲酸水溶液(8∶92)为流动相,柱温为30 ℃,流速为1.0 mL/min,电喷雾离子源(ESI),负离子模式,m/z范围为50~600,样品分流进入质谱,进入质谱的流速为0.3 mL/min,干燥气流量为8 mL/min,毛细管温度为325 ℃,碰撞电压为25 V。
2.2 样品溶液制备
分别在强酸(1 mol/L HCl,60 ℃持续1 h)、强碱(1 mol/L NaOH,60 ℃持续1 h)、高温(湿热环境和干热环境分别为90 ℃和120 ℃,分别持续3 h和0.5 h)、氧化(3% H2O2,25 ℃,持续0.5 h)、光照(4 500±500 lx,25 ℃,1个月)条件下,对TP供试品溶液(1.0 mg/mL)进行强制降解试验。
2.3 有关物质的定性分析
TP供试品溶液强制降解实验的色谱图如图2所示,共检测到7种有关物质(RS 2、3、4、5、6、7和8),其保留时间分别为4.78、5.75、14.32、15.58、26.27、27.50和33.51 min。其中,RS 5和7为TP水解和聚合产物,二者均有对照品可供判断,但注射用硫普罗宁中其他有关物质以前从未报道过,也没有任何对照品可供参考。因此,根据Q-TOF中母离子和产物离子的信息,合理地推导了它们的结构。
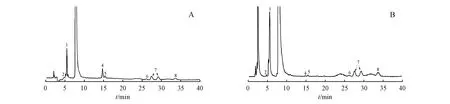
Figure2 Typical UV spectrum (A) and the total ion chromatogram (B) of forced degradation solution of TP for injection.The figures represent the RS (RS 2- 8)
根据图3,可以得出一些合理的RS转换途径。总结如下:RS 2是TP脱去巯基形成的,RS 3是TP与其降解产物结合形成的化合物,RS 4是TP的官能团异构体,RS 5是TP的水解产物,命名为2-巯基丙酸,RS 6是由TP的二聚体与其水解产物结合而形成的;RS 7是TP的二聚体,RS 8是由TP降解产物发生反应而形成的。
2.4 色谱条件
Thermo Scientific Syncronis C18柱(250 mm×4.6 mm,5 μm),流动相为乙腈-0.1%磷酸(8∶92),流速1.0 mL/min,柱温30 ℃,进样量20 μL,检测波长210 nm。
2.4.1 RS 7的测定 TP常以外消旋形式被用于临床,其结构中具有一个手性中心,二聚体产物RS 7就有两个手性中心,因此有3个不同的异构体,如图1-d所示。这3种异构体可分为两组对映体。在本研究中,RS 7分别在22和23 min出现双峰,面积比为1∶1。这两个峰是RS 7的两组对映体,用目前的色谱系统可以达到部分分离(分离度为1.1)。因此,在本研究中根据两个峰的总面积来计算RS 7的含量。
2.4.2 色谱条件的优化-流动相pH的优化 在4.0~1.5范围内对流动相pH进行了考察,在pH 4.0时,TP不能与中间产物RS 1达到完全分离(图4),主要原因是在较高的pH条件下,巯基不能保持足够的稳定性。随着流动相pH从4.0降至1.5,TP的拖尾因子由0.60逐渐提高到1.05,理论塔板数也由2 312显著增加到14 491。其主要原因是在pH较低的环境中TP解离受到抑制,从而使拖尾峰消失,柱效提高。
除此之外,TP的峰形也发生了变化(图5)。考虑到TP是半胱氨酸类似物,根据文献[19],半胱氨酸的pKa分别为1.71(-COOH),8.18(SH)或8.37(-SH),而色谱峰发生变形和分裂的原因是当流动相pH(pH 2.0)接近TP的pKa时,羧基解离和未解离状态的量级相等,同时,它也可能伴随着巯基和硅醇基的相互作用。随着pH的进一步降低,这种情况受到抑制,TP的峰形明显改善。
pH和pKa对TP拖尾因子和塔板数的影响,在一定程度上解释了TP及其杂质在不同pH条件下具有不同的保留时间以及在pH 2.0时主峰发生分裂的原因。当流动相pH从4.0下降到3.0时,TP和RS 7在色谱柱上的保留增强,TP的保留时间从8 min推迟到12 min,RS 7的保留时间从15 min推迟到43 min。有趣的是,当流动相pH降低到1.5时,TP和RS 7的保留时间分别向9和24 min靠近。其原因可能与TP和RS 7的pKa有关。用ACD/Labs预测它们的pKa分别为3.4和3.0。由此推测,当pH从4.0降到3.0时,TP和RS 7的羧基都是游离态的,分子间作用力增强,从而加强了它们的保留。当pH降至1.5时,TP和RS 7的酰胺基结合氢离子,使保留减弱,色谱峰向前移。
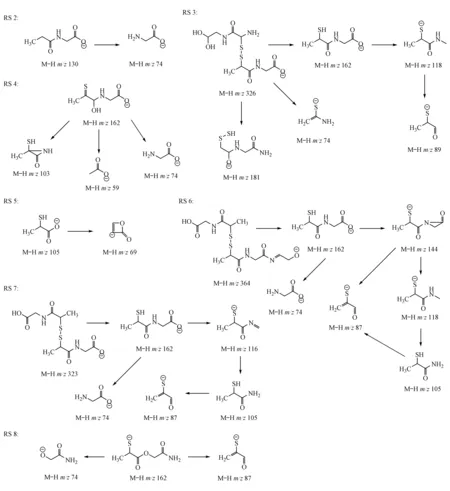
Figure3 Plausible pathway showing the formation of product ions of RS 2 (m/z130),RS 3 (m/z326),RS 4 (m/z162),RS 5 (m/z105),RS 6 (m/z364),RS 7 (m/z323) and RS 8 (m/z162)

Figure4 Chromatograms of system suitability solutions for different pH conditions with detection wavelength at 210 nm.The concentration of four reference substances:TP,RS 1,RS 5 and RS 7 were 100,10,10 and 15 μg/mL,respectively

Figure5 Chromatograms of sample solution at different pH conditions (pH 4.0- 1.5) with detection wavelength at 210 nm
除了上述提到的那些现象之外,RS 2也表现出类似的峰形、柱效和保留时间的改变。在pH 4.0时,其保留时间为3.8 min,柱效较低(理论塔板数5 490)。在pH降低后,色谱柱对RS 2的保留增加,RS 2的保留时间从3.8 min延迟到4.1 min。在pH 1.5时,RS 2的保留时间又向前移动到3.8 min。这些结果表明,当使用RP-HPLC分离巯基类化合物和甘氨酸类似物时,较高的流动相pH会影响柱效,并可导致峰分裂。在低pH(约pH 1.5)的情况下,可以避免这些情况的发生,并提高柱效和分离度。
2.4.3 检测波长选择 由于TP及其大部分杂质只在200~230 nm有末端吸收,而且没有明显的吸收峰。为了保证尽可能多的杂质能够被检测到并具有良好的灵敏度,在消除末端吸收和保证目标化合物具有强紫外吸收的情况下,检测波长设置为210 nm。
2.5 溶液配制
2.5.1 TP、RS 1、RS 5和RS 7对照品储备液 分别精密称取TP、RS 1、RS 5和RS 7对照品适量,用流动相分别溶解并稀释制成每毫升约含TP、RS 1、RS 5和RS 7 100 μg的对照品储备液,-20 ℃贮存。
2.5.2 TP供试品溶液 精密称取TP对照品适量,用流动相溶解并稀释制成每毫升约含TP 1 mg的溶液。
2.5.3 系统适用性溶液 精密量取TP、RS 1、RS 5和RS 7对照品储备液适量,用流动相稀释并制成含每毫升含TP、RS 1、RS 5和RS 7分别为100、10、10和15 μg的溶液。
2.6 方法学验证
2.6.1 系统适用性 取系统适用性溶液,按照“2.4”项下色谱条件进样测定,记录色谱图。在该色谱条件下,TP与各杂质以及各杂质之间的分离度和理论塔板数最小分别为6.8(≥2.0)和15 057(≥2 000)。峰面积和保留时间的相对标准偏差分别为0.64%和0.06%,均小于1.0%。拖尾因子在1.01~1.07之间,小于2.0。所有数据均符合验收标准。由此可以看出,所建立的高效液相色谱系统适用于该分析方法。
2.6.2 专属性 按照“2.2”项下的条件对TP供试品溶液进行强制降解试验,并按“2.4”项下色谱条件进样测定,记录色谱图。当样品受到强酸、强碱、高温、光照和氧化破坏时,会出现一些新的色谱峰,可与TP完全分离。最小分离度为1.9(≥1.5)。此外,在所有破坏试验中,TP主峰纯度均合格,最小峰纯度指数大于零,表明该峰不含杂质。因此,该方法测定注射用硫普罗宁中的有关物质具有专属性。
2.6.3 回收率 取9份样品,每份分别精密加入相当于RS 1、RS 5和RS 7限度浓度的50%、100%和150%的RS对照品,配制低、中、高3种浓度的回收率溶液(n=9),进样测定。TP、RS 1、RS 5和RS 7的平均回收率分别为100.03%(RSD为1.02%)、101.96%(RSD为0.58%)、102.41%(RSD为2.60%)和100.40%(RSD为0.71%)。
2.6.4 精密度 取注射用硫普罗宁自制制剂,分别加入RS 1、RS 5和RS 7的对照品,制成含限度浓度杂质的模拟样品;按“2.4”项下色谱条件连续进样6次,记录色谱图。结果见表1。RS日内精密度和日间精密度最大的RSD分别为1.69%和4.42%(≤5.0%)。结果表明,该方法具有较高的精密度。
2.6.5 检测限(LOD)和定量限(LOQ) 将TP、RS 1、RS 5、RS 7的对照品储备液逐步稀释,按“2.4”项下色谱条件进样测定,记录色谱图,以S/N≥10得到TP、RS 1、RS 5、RS 7的定量限,以S/N≥3得到TP、RS 1、RS 5、RS 7的检测限(见表1),从表中可以看出,TP和杂质(RS 1、RS 5和RS 7)的LOD和LOQ分别在0.102 0~0.399 1 μg/mL和0.309 2~1.197 μg/mL范围内。根据注射用硫普罗宁的杂质限度,LOD和LOQ可分别达到0.01%和0.03%。结果表明,该方法对注射用硫普罗宁中的任何杂质均有足够的灵敏度。
2.6.6 线性和范围 分别精密量取TP、RS 1、RS 5、RS 7的对照品储备液,用流动相逐级稀释,分别得到0.30~7.5 μg/mL (0.30,1.5,3.0,5.0,6.0和7.5 μg/mL),0.30~7.5 μg/mL (0.30,1.5,3.0,5.0,6.0和7.5 μg/mL),1.2~7.5 μg/mL(1.2,2.4,3.6,5.0,6.0和7.5 μg/mL)和1.2~50 μg/mL(1.2,5.0,15,25,40和50 μg/mL)范围内的标准系列浓度溶液,按“2.4”项下色谱条件进样测定,记录色谱图,以峰面积和质量浓度(μg/mL)分别表示为Y和X,进行线性回归,其线性方程见表1。结果表明,在不同浓度范围内各杂质的线性关系良好。所有结果都符合标准。
2.6.7 耐用性 通过改变流速(±0.1 mL/min)、柱温(±5 ℃)、波长(±2 nm)、流动相中乙腈含量(±1%)和同一品牌不同批次分析柱等试验条件,考察该方法的耐用性。实验结果(见表2)。从表中可知,RSD为0.09%~6.41%,说明该方法具有足够的耐用性。
Table1 Validation results of introduced method for determination of related substances of tiopronin for injection

AnalyteLinearrange/(μg/mL)Regressionequation(x±s,n=3)rLOD/(μg/mL)LOQ/(μg/mL)Inter-dayprecisionRSD/%(n=6)Inter-dayprecisionRSD/%(n=12)TP0.30-7.5y=14961x-4470.99990.10200.30920.120.83RS10.30-7.5y=13818x-6540.99980.10320.31281.601.91RS51.20-7.5y=7108x-160.99990.38801.1641.691.41RS71.20-50y=21455x-44380.99980.39911.1970.864.42
aLow:80% level of limitation;bMiddle:100% level of limitation;cHigh:120% level of limitation


AnalyteFlowrate(±0.1mL/min)Columntemperature(±5℃)λ(±2nm)Acetonitrilecontent(±1%)DifferentcolumnTP101.0±0.1100.8±0.4101.1±0.2100.9±0.3101.0±0.3RS1100.3±0.6100.2±4.2100.0±4.8102.0±1.2103.4±0.9RS599.4±2.3101.7±5.899.8±6.4102.9±2.5101.5±2.0RS7104.2±4.7101.1±1.0100.1±1.0100.8±5.6100.6±3.0
2.6.8 溶液稳定性 将RS 1、RS 5和RS 7的对照品溶液在室温下放置24 h(时间点分别为0、1、2、4、6、8、10、16和24 h),其峰面积的RSD(%)分别为0.11、0.30和0.19,在4 ℃冰箱中放置24 h(时间点分别为0、1、2、4、6、8、10、16和24 h),其峰面积的RSD(%)分别为0.33、0.35和0.30。TP对照品溶液在室温下放置24 h(时间点分别为0、2、4和24 h),其峰面积的RSD(%)分别为0.43,在4 ℃冰箱中放置24 h(时间点分别为0、2、4、11、18和24 h),其峰面积的RSD(%)为0.43。样品溶液分别置于室温和4 ℃冰箱中24 h(时间点分别为0、2、5、9、12、15、18、21和24 h)后,峰面积RSD(%)分别为0.15和0.23。实验结果表明,对照品溶液和样品溶液至少在24 h内是稳定的。
2.7 样品测定
根据HPLC方法的建立和验证,对13批注射用硫普罗宁样品的RS进行了研究。其中,RS 1作为合成过程中的一种可能的中间副产物并没有被检测到。同时,本研究还发现未知杂质中的主要杂质(RS 2)在每一批中都有规律的再现。由于缺少RS 2的对照品,所以用自稀释法计算其含量。注射用硫普罗宁的RS 2、RS 5、RS 7和总杂含量分别小于0.15%、0.07%、1.8%和2.0%。
3 讨论与小结
本研究建立了一种新的、简便的测定注射用硫普罗宁的反相高效液相色谱方法,并对其进行了验证。该分析过程揭示了巯基化合物和甘氨酸类似物在反相高效液相色谱上的保留以及流动相pH对待测物色谱行为的影响。用该方法共检测到7种有关物质,并用LC-Q-TOF对其进行了表征,包括过程工艺杂质和降解产物,其中5种尚未见文献报道。由于流动相组成简单,实验装置通用性强,使该方法成为分析测定注射用硫普罗宁的首选方法,并为国内外药典对注射用硫普罗宁标准的制定提供了重要参考。
猜你喜欢
理化检验-化学分册(2022年10期)2022-10-21 08:07:30
中西医结合心脑血管病杂志(2022年12期)2022-07-07 10:24:24
中华养生保健(2020年2期)2020-11-16 00:49:20
云南中医学院学报(2015年1期)2015-07-31 18:10:45
四川师范大学学报(自然科学版)(2015年1期)2015-02-28 14:07:29
海洋科学进展(2015年1期)2015-02-27 13:16:16
中国现代药物应用(2015年8期)2015-01-23 12:36:27
中国药业(2014年17期)2014-05-26 09:07:49
食品科学(2013年23期)2013-03-11 18:30:02
红河学院学报(2012年4期)2012-12-27 12:07:10
