
拉洛他赛的波谱解析及高效液相色谱法对其脂质体含量的测定
2019-06-18 10:25李雪琦李建伟李秋红段嘉伦崔一诺苏展博许佳瑞杜亚菲王桂玲吕万良
北京大学学报(医学版) 2019年3期
李雪琦,李建伟,2,3,4,李秋红,4,阎 妍,段嘉伦,崔一诺,苏展博,罗 倩,许佳瑞,杜亚菲,王桂玲,谢 英,吕万良△
(1. 天然药物与仿生药物国家重点实验室,分子药剂学与新释药系统北京市重点实验室,北京大学药学院, 北京 100191; 2. 北京大学前沿交叉学科研究院, 北京 100871; 3. 山西振东制药股份有限公司制剂所, 山西长治 047100; 4. 山西大学中医药现代研究中心, 太原 030006)
拉洛他赛(larotaxel, XRP9881, RPR109881)最初是由Sanofi-Aventis公司开发的通过半合成方法得到的一种新型的紫杉烷类细胞周期特异性化疗药,具有较广的抑瘤谱。目前为止,该药在国内外均未上市,我国山西振东药业正在进行拉洛他赛脂质微球注射剂的新药开发,并获得了国家食品药品监督管理局批准,目前正开展临床试验研究。
与其他紫杉烷类化疗药相似,拉洛他赛可作用于细胞分裂过程中纺锤体微管蛋白,与游离的微管蛋白结合,促进微管蛋白装配成稳定微管并抑制微管解聚,从而抑制癌细胞的有丝分裂。初步研究显示,拉洛他赛区别于其他紫杉烷类抗肿瘤药,其可杀伤耐药性癌细胞,还可透过血脑屏障,表现出很好的抗肿瘤应用前景[1-4]。体外研究表明,拉洛他赛的抗肿瘤活性强于紫杉醇,对多药耐药性肿瘤细胞展现出较强的抑制和杀伤作用[5-7]。现有的临床研究结果表明,拉洛他赛作为二线治疗或补救治疗药物,治疗转移性乳腺癌的疗效优于多西他赛和多柔比星[8-10]。
有文献报道显示,国外用于临床试验的拉洛他赛制剂中,一般加入表面聚山梨酯作为增溶剂,以增加拉洛他赛的溶解度[11],例如Sanofi-Aventis公司在用于二期临床试验的拉洛他赛注射剂中采用了聚山梨酯(吐温-80)作为增溶剂。为避免采用表面活性剂可能导致的患者过敏,增强拉洛他赛对肿瘤组织的被动靶向性聚集,本课题组研究了一种拉洛他赛脂质体[12]。
为了对拉洛他赛及其脂质体制剂进行全面的定性和定量表征,本研究采用红外吸收光谱、核磁共振波谱(nuclear magnetic resonance spectroscopy, NMR)、质谱和紫外光谱,对拉洛他赛的分子结构进行了光谱学解析,同时建立一种采用高效液相色谱(high performance liquid chromatography,HPLC)对拉洛他赛进行含量测定的方法,为建立拉洛他赛制剂的质量标准提供依据。
1 材料与方法
1.1 实验材料
拉洛他赛(larotaxel)和拉洛他赛对照品(纯度:99.8%)由山西振东制药有限公司提供,氘代氯仿购自北京百灵威科技有限公司,甲醇(HPLC级)和乙腈(HPLC级)购自Burdick & Jackson公司(美国),二棕榈酰磷脂酰胆碱(1,2-dipalmitoyl-sn-glycero-3-phosphocholine,DPPC)、二硬脂酰磷脂酰乙醇胺-聚乙二醇(distearoylphosphatidyl ethanolamine-polyethylene glycol, DSPE-PEG2000)购自Avanti公司(美国),胆固醇购自北京市海淀区微生物培养基制品厂,其余试剂均为分析纯,购自北京国药试剂公司。
1.2 实验仪器
BT-25S精密电子天平(十万分之一)购自德国Sartorius公司,安捷伦1260型高效液相色谱仪购自Agilent公司(美国),ZORBAX SB-C18反相色谱柱(Agilent, 5 μm, 4.6 mm×250 mm)购自Agilent公司(美国),红外光谱仪(Nexus 470)购自Nicolet公司(美国),核磁共振波谱仪(Avance III 400)购自Bruker公司(德国),高分辨质谱(Xevo G2 Q-TOF)购自Waters公司(英国),紫外可见分光光度计(UV-1800型)购自上海美谱达仪器有限公司,RE52CS旋转蒸发仪购自上海亚荣生化仪器厂,SB-5200超声机购自宁波新芝生物科技股份有限公司,JY92-IID型超声波细胞粉碎机购自宁波新芝生物科技股份有限公司,激光散射粒径测定仪(Malvern zetasizer 3000HS)购自Malvern公司(英国),透射电镜(Tecnar G2 20ST型,TEM)购自FEI公司(美国)。
1.3 拉洛他赛的四大光谱测定
1.3.1质谱测定 采用单四级杆质谱仪(quadrupole mass spectrometer),以甲醇作为液相基质对拉洛他赛进行检测。
1.3.2红外吸收光谱测定 采用KBr压片法将拉洛他赛固体粉末进行压片,然后检测。分辨率为4.00,采样增益为1.0,动镜速度为0.474 7,光阑为100.00,检测器为DTGS KBr,分束器为KBr,光源为红外光源。
1.3.3核磁共振谱测定 将10.0 mg拉洛他赛溶于0.5 mL CDCl3,然后置于核磁管进行检测,检测温度恒定为25 ℃。核磁共振谱包括:1H NMR,13C NMR,1H-1H COSY(1H-1H chemical-shift correlation spectroscopy), HSQC(heteronuclear single quantum correlation),HMBC(heteronuclear multiple bond correlation)和DEPT(distortionless enhanced polarization transfer)135光谱。核磁化学位移为:CDCl3(TMS,1H NMRδ0.00,13C NMRδ0.0 ppm,ppm为百万分之一相对化学位移)。
1.3.4紫外吸收光谱测定 取适量0.25 g/L的拉洛他赛标准溶液,在200~800 nm波长范围内进行光谱扫描。
1.4 拉洛他赛高效液相色谱法的建立
1.4.1色谱条件 色谱柱为 SB-C18 柱(5 μm, 250 mm×4.6 mm),测定温度为25 ℃,流动相为乙腈-水(体积比为75 ∶25),流速为1.0 mL/min,进样量为10 μL,检测波长为230 nm。
1.4.2标准曲线 精密称取拉洛他赛对照品适量,加入甲醇溶解,制备200.0 mg/L的拉洛他赛贮备液,并倍比稀释成0.5、1.5、3.0、6.0、12.0、25.0、50.0、100.0 mg/L系列浓度的拉洛他赛溶液样品。按照上述色谱条件,取不同浓度的溶液分别进样,用高效液相色谱仪检测每一浓度的峰面积A值,并以浓度C为自变量,对相应的色谱峰面积A进行线性回归,求得拉洛他赛的标准曲线。
1.4.3稳定性 精密称取拉洛他赛对照品适量,加入甲醇溶解,制备成5.0、10.0和25.0 mg/L溶液,于制备后0、2、4、6、8 h分别用上述色谱条件测定峰面积,平行测定3次,取其平均值并计算相对标准偏差。
1.4.4回收率 取适量的空白脂质体,加入9倍体积的甲醇使脂质体破坏和溶解,加入精密称定的拉洛他赛对照品溶液适量,以甲醇为溶剂,配制成 5.0、10.0和25.0 mg/L溶液各3份,分别用上述色谱条件测定峰面积,计算回收率和相对标准偏差。
1.4.5精密度 取适量的空白脂质体,加入9倍体积的甲醇使脂质体破坏和溶解,加入精密称定的拉洛他赛对照品适量,以甲醇为溶剂,配制成5.0、10.0和25.0 mg/L拉洛他赛的系列浓度溶液,于5 d内分别用上述色谱条件测定峰面积,计算日内和日间相对标准偏差。
1.4.6检测限 精密称定的拉洛他赛对照品适量,加甲醇溶解并稀释,按倍比配制系列浓度,分别按照上述色谱条件测定样品的色谱峰,当信噪比约为3 ∶1时,其检测浓度被认为是本方法的检测限。
1.5 拉洛他赛脂质体的制备与表征
1.5.1拉洛他赛脂质体的制备 精密称取DPPC、胆固醇、DSPE-PEG2000(60 ∶40 ∶5, μmol/μmol)和拉洛他赛(质量比为药 ∶脂材=1 ∶20)置于茄形瓶中,加入适量氯仿 ∶甲醇(体积比为3 ∶1)溶液溶解,然后在40 ℃水浴减压蒸发除去有机试剂,在瓶底和内壁形成一层均匀的脂膜。加入适量磷酸盐缓冲液(pH 6.8)水化,先在水浴中超声2~3 min,形成乳白色均匀的粗脂质体,然后转移到超声波细胞粉碎机中进行探头超声,设置超声工作时间为1 s,间歇时间为1 s,全程时间为10 min,保护温度为35 ℃,功率为200 W。探头超声结束后,形成带有弱蓝色乳光的半透明液体,再将所得脂质体依次挤压通过孔径为400 nm、200 nm的聚碳酸酯膜,各挤压过膜3次即得到拉洛他赛脂质体。
1.5.2空白脂质体的制备 与拉洛他赛脂质体的制备方法相同,但在成膜过程中不加入拉洛他赛。
1.5.3拉洛他赛脂质体的形态观察 以去离子水作为分散介质,适当稀释拉洛他赛脂质体,将稀释后的拉洛他赛脂质体液再经200 nm的微孔滤膜过滤,然后将铺有碳膜的铜网漂放在脂质体溶液上,1 min后取出用滤纸吸干,将俘获有脂质体粒子的铜网漂放在1%(体积分数)醋酸双氧铀水溶液上,1 min后取出用滤纸吸干,静置过夜,置于透射电镜下观察。
1.5.4拉洛他赛脂质体粒径和Zeta电位的测定 取新制得的拉洛他赛脂质体各1 mL,加磷酸盐缓冲溶液(pH 6.8)适当稀释,利用激光散射粒径测定仪测定。测定温度设定为25 ℃,每个样品测定20次,记录其粒径、多分散系数和Zeta电位值并计算平均值。
1.5.5拉洛他赛在脂质体中包封率的测定 取新制备的拉洛他赛脂质体500 μL,使之通过Sephadex G-50葡聚糖凝胶柱,以磷酸盐缓冲溶液(pH 6.8)为流动相,分离未包封于脂质体的游离拉洛他赛,收集分离后的脂质体,加甲醇破坏,然后用高效液相色谱法进行测定。另外,取未过凝胶柱的拉洛他赛脂质体,适当稀释后加入流动相破坏,然后用上述高效液相色谱法进行测定。拉洛他赛在脂质体中的包封率用以下公式计算:包封率=过凝胶柱脂质体中测得拉洛他赛量/未过凝胶柱脂质体中测得拉洛他赛量×100%。按照外标法,平行测定拉洛他赛对照溶液中的药物量作为参比标准,计算拉洛他赛在脂质体中的包封率。
2 结果
2.1 拉洛他赛的质谱测定结果
拉洛他赛的正离子模式高分辨质谱见图1A,结果显示,测得m/z854.336 6为[M+Na]+(计算值为854.335 8),m/z832.354 9为[M+H]+(计算值为832.408 0)。拉洛他赛的负离子模式高分辨质谱见图1B,结果显示,测得m/z876.345 4为[M+2Na-H]-(计算值为876.335 8),m/z830.339 0为[M-H]-(计算值为830.408 0)。根据两种模式下的质谱结果,可确定其分子式为C45H53NO14,其相对分子质量为831.900 1。
2.2 拉洛他赛的红外吸收光谱测定结果

2.3 拉洛他赛的核磁共振光谱测定结果
拉洛他赛的核磁共振碳谱和氢谱分别见图3A、3B。拉洛他赛的二维核磁共振谱分别见图4A(1H-1H COSY)、4B(HSQC)、4C(HMBC)和4D(DEPT 135)。
拉洛他赛的一维核磁共振谱中共出现45个碳信号和53个质子信号,其中C-19为拉洛他赛的特征结构,其化学位移为15.63,所对应的两个质子的化学位移分别为2.25 (1H, m)、1.66 (1H, t,J=5.8 Hz)。
拉洛他赛的二维核磁共振谱结果显示,1H-1H COSY二维谱可以见到相耦合的质子相关信号,HSQC二维谱中可见到各个碳与相应质子的相关信号,HMBC二维谱中可见各个碳与相邻质子的相关信号,DEPT 135二维谱所得碳的分类与结构式基本一致,共8个CH3、4个CH2、18个CH和15个季碳。
经1H-1H COSY、HSQC、HMBC和DEPT二维波谱验证,1H NMR(400 MHz,CDCl3)和13C NMR(125 MHz,CDCl3)波谱提供的质子信号和碳信号全部进行归属[13],详细结果见表1,化学结构见图5。
综合上述一维及二维NMR谱所给出的信号,可以给全部碳、氢信号做出正确归属,证实拉洛他赛的化学结构式准确无误,相应的,本研究所测定绘制的谱可望作为拉洛他赛参考标准核磁共振波谱。
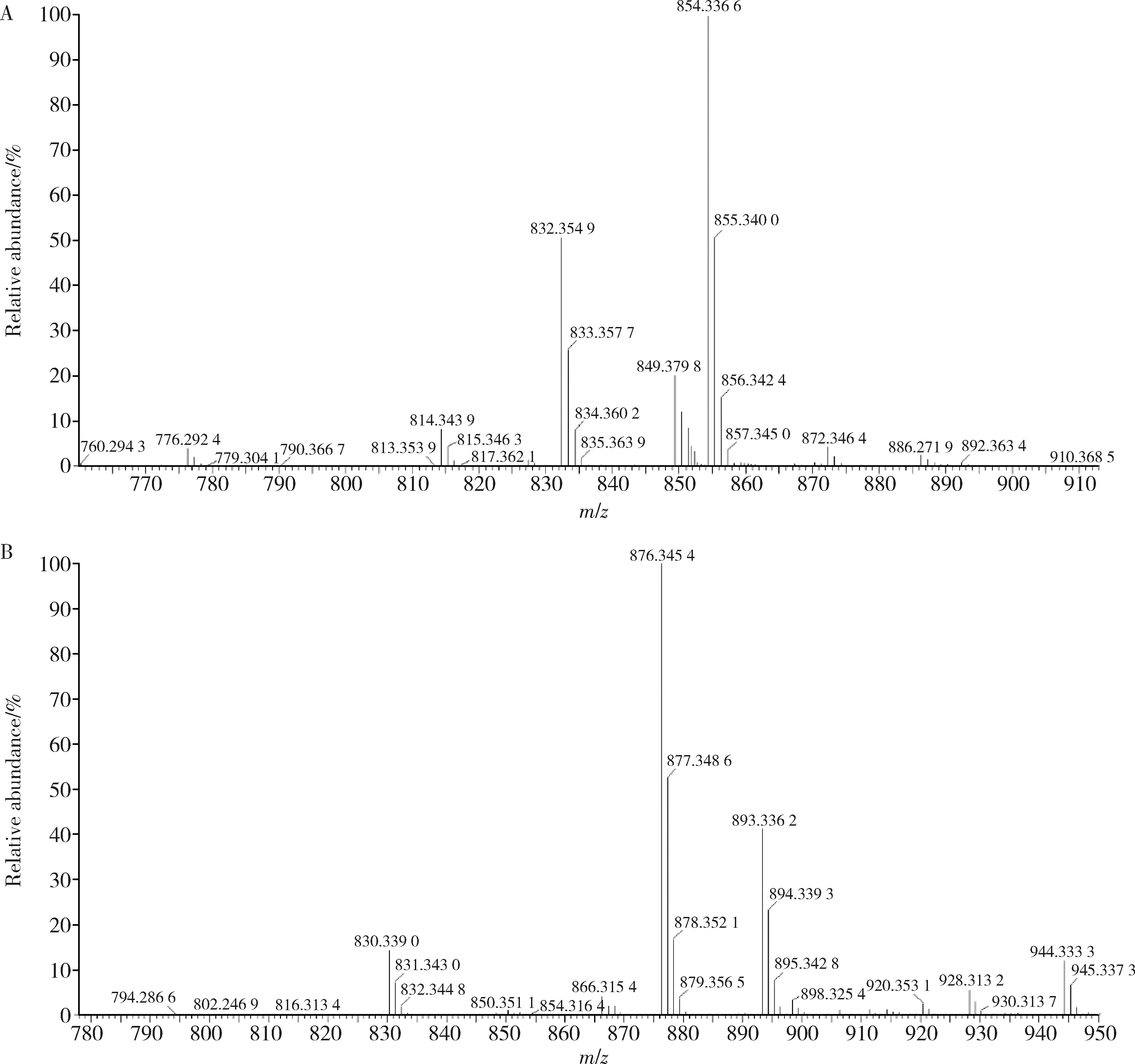
A, positive ion mode; B, negative ion mode.图1 拉洛他赛的高分辨飞行时间质谱图Figure 1 Mass spectrums of larotaxel by quadrupole-time of flight mass spectrometry (Q-TOF-MS)
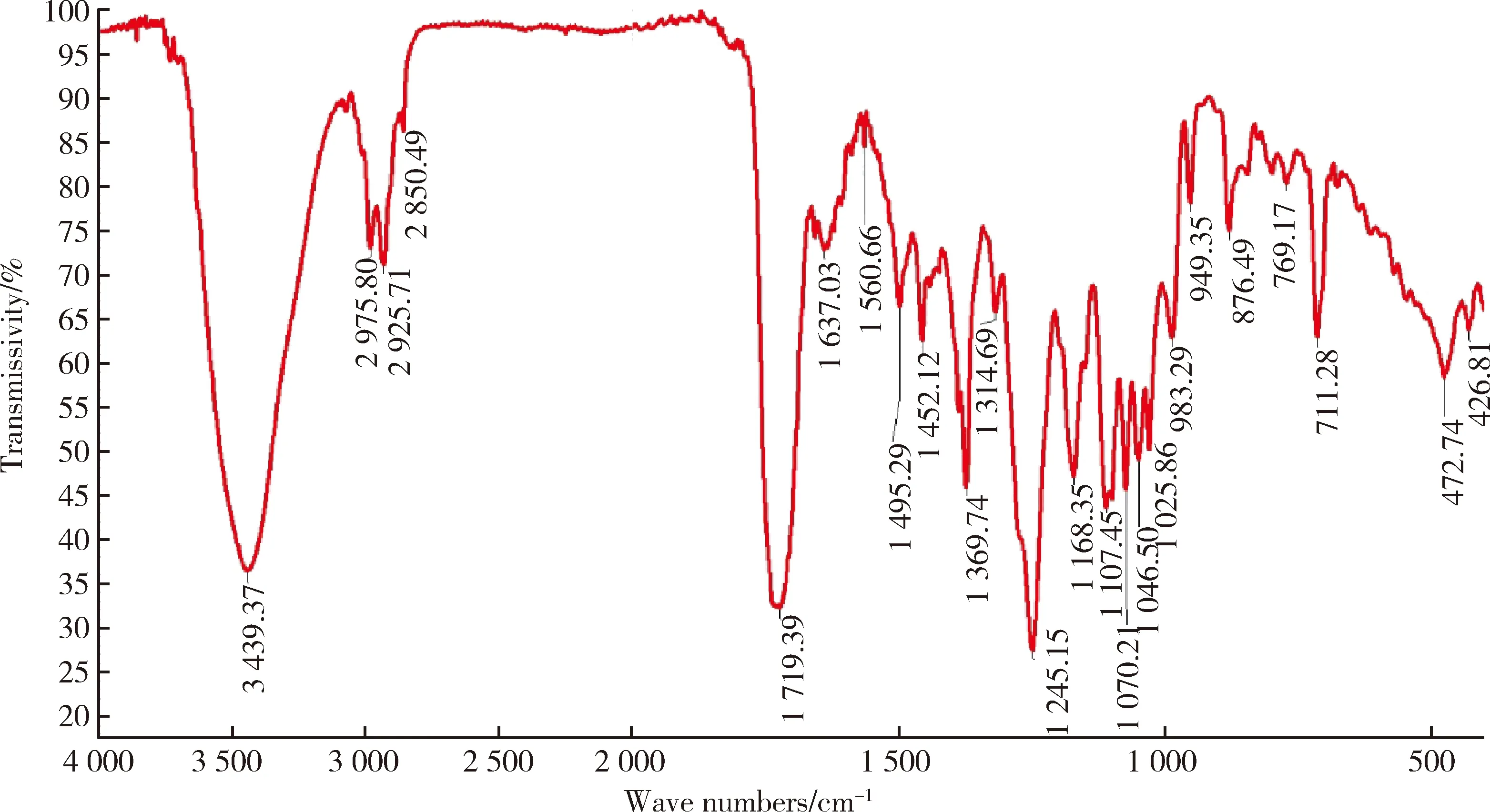
图2 拉洛他赛的红外吸收光谱Figure 2 Infra-red absorption spectrum of larotaxel

A, 13C NMR spectrum of larotaxel; B, 1H NMR spectrum of larotaxel. NMR, nuclear magnetic resonance; ppm, one millionth chemical shift.图3 拉洛他赛的核磁共振碳谱及氢谱Figure 3 13C NMR and 1H NMR spectrums of larotaxel
2.4 拉洛他赛的紫外-可见吸收光谱扫描结果
当拉洛他赛甲醇溶液为0.25 g/L时,将该溶液在200~800 nm波长范围内进行光谱扫描,得到拉洛他赛的紫外-可见吸收光谱图(图6)。根据其吸收光谱可知,拉洛他赛在203 nm处有最大吸收,在230 nm附近有较强的紫外吸收信号,但是250 nm处吸收信号很弱,在随后波长的紫外和可见区拉洛他赛没有吸收值。
2.5 拉洛他赛的高效液相色谱测定方法
2.5.1标准曲线 按照上述色谱条件,测定系列浓度的拉洛他赛溶液样品的色谱峰面积A值,并以浓度C(mg/L)为自变量,对其相应的色谱峰面积A值进行线性回归,求得标准曲线(图7A)。拉洛他赛的标准曲线为A=11.82C+0.01,R2=0.999 9,具有很好的线性关系。拉洛他赛在此液相条件下的标准色谱图见图7B,拉洛他赛色谱峰的保留时间为5.71 min左右,峰型规整、对称。
2.5.2稳定性 按照上述色谱条件,对浓度为 5.0、10.0和25.0 mg/L的拉洛他赛甲醇溶液分别在0、2、4、6、8 h时测定峰面积(表2),结果表明,拉洛他赛在8 h内测定结果基本稳定,相对标准偏差(relative standard deviation,RSD)值小于2.0%。由此可知,拉洛他赛的甲醇溶液在8 h内具有较好的稳定性,进行含量测定时无需新鲜配制。

A,1H-1H chemical correlation NMR (1H-1H COSY) spectrum; B, 1H detected heteronuclear singular quantum correlation (HSQC) NMR spectrum; C, 1H detected heteronuclear multiple bond correlation (HMBC) NMR spectrum; D, distortionless enhanced polarization transfer (DEPT) 135 NMR spectrum. NMR, nuclear magnetic resonance; ppm, one millionth chemical shift.图4 拉洛他赛的二维核磁共振谱Figure 4 2D-NMR spectrums of larotaxel
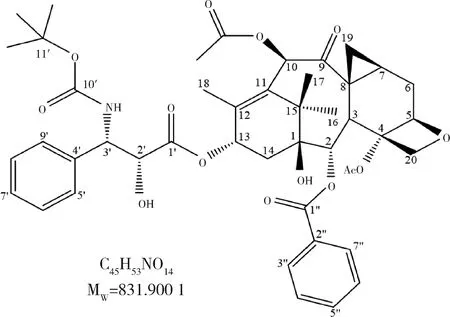
图5 拉洛他赛的结构式和相对分子质量Figure 5 The structural formula and the relative molecular mass of larotaxel
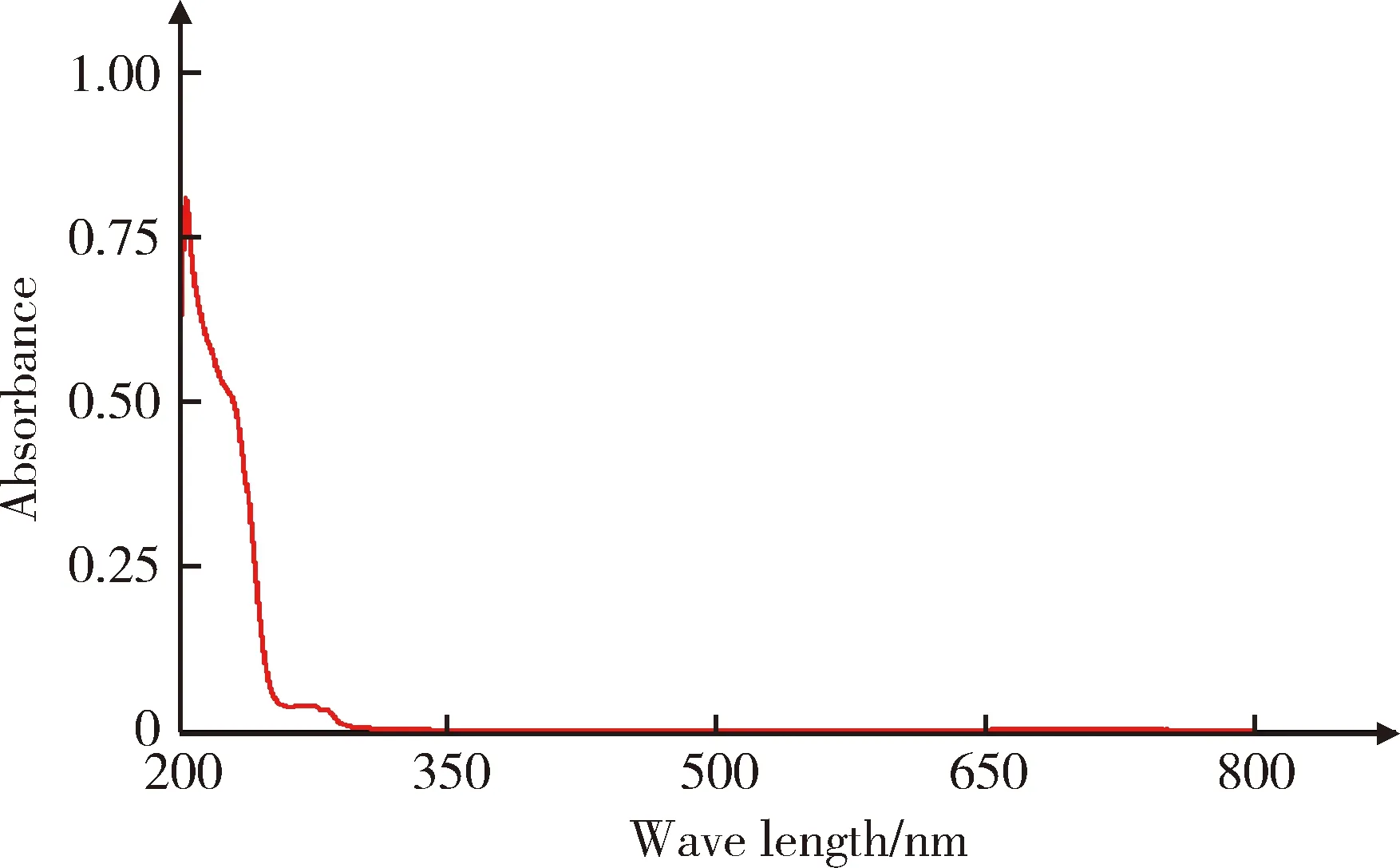
图6 拉洛他赛甲醇溶液(250 mg/L)在200~800 nm波长范围内的紫外吸收扫描光谱Figure 6 Ultra-violet visible spectrophotometric scanning spectrum of larotaxel methanol solution (250 mg/L) in the range of 200-800 nm
表1 拉洛他赛的13C NMR和1H NMR波谱解析数据Table 1 1H NMR and 13C NMR assignments for larotaxel

s, singlet; d, doublet; t, triplet; m,multiplet; dt, double triplet; br.s, broad singlet; br.d, broad doublet. NMR, nuclear magnetic resonance.

Column: SB-C18 (5 μm, 250 mm×4.6 mm); mobile phase: acetonitrile-water (75 ∶25, volume/volume); detection wavelength: 230 nm. A, correlation profile between concentration and peak area values of larotaxel measured by HPLC-UV method; B, typical chromatogram of laro-taxel measured by HPLC-UV spectrum; C, chromatogram of limit of quantitation of larotaxel measured by HPLC. UV, ultraviolet; HPLC, high performance liquid chromatography.图7 高效液相-紫外色谱法测定拉洛他赛的浓度与色谱峰面积相关曲线及典型色谱图Figure 7 Correlation profile between concentration and peak area values of larotaxel measured by HPLC-UV method and typical chromatographs
2.5.3回收率 使用上述色谱条件对3份浓度分别为5.0、10.0和25.0 mg/L的拉洛他赛溶液进行测定(表3),结果表明,拉洛他赛对照品的回收率为100%±2%,RSD值均小于1%,脂质体制剂中拉洛他赛的回收率为100%±3%,RSD值均小于2%。此含量测定方法对拉洛他赛对照品和脂质体制剂中的拉洛他赛均具有很好的准确性。
2.5.4精密度 使用上述色谱条件,于5 d内对5份浓度分别为5.0、10.0和25.0 mg/L溶液进行测定(表4),数据显示,拉洛他赛对照品的精密度为100%±5%,RSD值均小于2%,脂质体制剂中拉洛他赛的精密度为100%±5%,RSD值均小于2%。此含量测定方法对拉洛他赛对照品和脂质体制剂中的拉洛他赛含量测定均具有较好的精密度。
2.5.5检测限 采用高效液相色谱法测量拉洛他赛的检测限约为50.0 μg/L,在检测限时的色谱图见图7C。
2.6 拉洛他赛脂质体的制备与表征结果
制备的拉洛他赛脂质体带有微弱淡蓝色乳光,其透射电镜图显示,该脂质体呈圆球形的囊状结构(图8)。在该脂质体中,由于拉洛他赛是脂溶性药物,与脂质体膜材一起溶解然后成膜,故拉洛他赛被包载于磷脂双分子层中。
激光粒度仪分析结果显示(表5),拉洛他赛脂质体的平均粒径为(105.73±2.30) nm,多分散系数为0.190±0.072,zeta电位为(0.240±0.015) mV。
高效液相色谱法测定该脂质体中拉洛他赛的包封率为87.73%±2.70%。

表2 拉洛他赛甲醇溶液HPLC-UV测定时的稳定性Table 2 HPLC-UV measurement stability of larotaxel in methanol
Data are presented as the mean±standard deviation (n=3). UV, ultraviolet; HPLC, high performance liquid chromatography; RSD, relative stan-dard deviation.

表3 拉洛他赛对照品及脂质体中用高效液相-紫外色谱法进行拉洛他赛含量测定的回收率Table 3 Recoveries of standard larotaxel and larotaxel in the liposomes measured by HPLC-UV
1, recoveries of standard larotaxel; 2, recoveries of larotaxel in the liposomes. Data are presented as the mean±standard deviation (n=3). Abbre-viations as in Table 2.
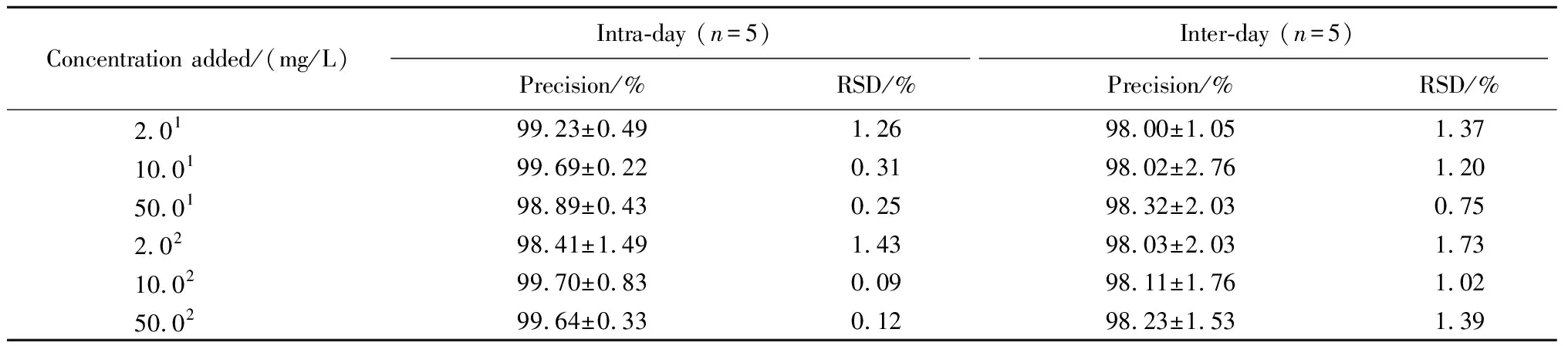
表4 拉洛他赛对照品及脂质体中用高效液相-紫外色谱法进行拉洛他赛含量测定的精密度Table 4 Precisions of standard larotaxel and larotaxel in the liposomes measured by HPLC-UV
1, recoveries of standard larotaxel; 2, recoveries of larotaxel in the liposomes. Data are presented as the mean±standard deviation (n=5). Abbre-viations as in Table 2.

表5 拉洛他赛脂质体的理化表征Table 5 Physicochemical characterization of larotaxel liposomes
Data are presented as the mean±standard deviation (n=3). PDI, polymer dispersity index.
3 讨论
鉴于拉洛他赛在抗耐药性和转移性乳腺癌方面的药效学优势,目前国内外都在积极开展临床试验研究。本研究利用高分辨质谱、红外吸收光谱、核磁共振谱和紫外-可见光吸收光谱对拉洛他赛分子式、相对分子质量及化学结构进行了确证与解析,提供的四大光谱图可作为拉洛他赛的参考标准图谱,用作其药物研究、药品鉴定、药品开发检验和生产质量控制等。
在正离子模式下的高分辨质谱图中,拉洛他赛呈现两个强信号峰,分别为m/z854.336 6和m/z832.354 9,分别对应[M+Na]+和[M+H]+;在负离子模式下的高分辨质谱图中,拉洛他赛呈现两个强信号峰,分别为m/z876.345 4和m/z830.339 0,对应[M+2Na-H]-和[M-H]-。两种模式下的质谱结果,验证了拉洛他赛的分子式为C45H53NO14,相对分子质量为831.900 1。
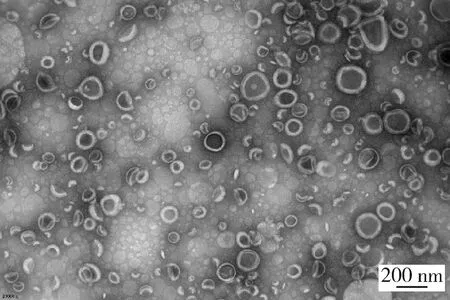
图8 拉洛他赛脂质体的透射电镜图像Figure 8 Transmission electron microscope (TEM) image of larotaxel liposomes
在拉洛他赛的红外吸收光谱中,化学结构中的苯环、酯基、羟基、叔丁基和酰胺结构等关键官能团均有相应吸收峰,因此进一步验证了其分子结构上的特征吸收峰,且吸收峰强度适宜、无杂峰,可以作为拉洛他赛的红外吸收光谱标准对照图谱,用于该药品的定性与鉴定。
拉洛他赛的核磁共振谱中的碳信号和质子信号较全面,信号强度适当并且几乎无杂质峰信号,在二维核磁共振谱的指导下,1H和13C信号均能得到合理归属,因此,呈现的核磁共振碳谱和氢谱均可作为拉洛他赛的标准核磁共振谱,用于其药品鉴定和纯度检测。
在拉洛他赛的紫外-可见光吸收光谱中,拉洛他赛在203 nm处有最强吸收,但是在该波长处紫外测定具有较强的溶剂干扰效应,且在其他波段无明显吸收,故采用常用的紫外-可见分光光度法无法对拉洛他赛进行定量测定。虽然如此,拉洛他赛在230 nm处有较强的紫外吸收,故230 nm可用作高效液相-紫外色谱检测的波长。
为此,我们进一步建立了高效液相-紫外色谱法,旨在建立拉洛他赛的定量测定方法。经方法有效性验证显示,该方法灵敏、高效,具有很好的线性关系,且回收率和精密度高,具备良好的稳定性和准确性,可用于拉洛他赛原料药和脂质体制剂中拉洛他赛的含量测定。
此外,本研究制备的拉洛他赛脂质体稍呈正电性、形态规整、粒径适当、大小分布均一、载药包封率高。初步观察显示,该脂质体不易发生聚沉,其脂质体膜上用含PEG的磷脂材料修饰,预期也可以增加给药后在血液系统中的稳定性,延长体内循环时间,减少网状内皮系统快速清除与代谢,从而增加拉洛他赛脂质体在肿瘤组织中的富集和滞留,有利于提高药效并降低全身副作用[14-16]。
综上所述,本研究对拉洛他赛进行了详细的高分辨质谱、红外吸收光谱、核磁共振谱和紫外-可见光谱测定,进行了波谱解析并制定了相应的参考标准图谱,验证并确认了拉洛他赛的分子式、相对分子质量和结构式,建立了拉洛他赛的高效液相-紫外色谱含量测定方法,可用于新药拉洛他赛脂质体的质量控制。
猜你喜欢
纺织标准与质量(2022年1期)2022-07-12
昆钢科技(2022年2期)2022-07-08
昆钢科技(2022年2期)2022-07-08
当代水产(2022年4期)2022-06-05
昆明医科大学学报(2022年3期)2022-04-19
昆钢科技(2021年4期)2021-11-06
口腔护理用品工业(2021年4期)2021-11-02
中国药学药品知识仓库(2021年18期)2021-02-28
信息技术时代·上旬刊(2019年4期)2019-09-10
分析化学(2018年12期)2018-01-22
