
基于R语言爬虫对Illumina接头序列的挖掘
2019-06-10 09:31柏程思
科技创新导报 2019年5期
柏程思
摘 要:当前生物信息学过滤测序接头序列的软件不能涵盖所有Illumina测序平台的接头序列。这样造成了分析NGS数据平台的局限性。本文通过R语言编程利用爬虫技术对Illumina质控文件的分析,挖掘所有不能被过滤软件识别的接头序列。
关键词:生物信息学 R语言编程 爬虫 Illumina測序
中图分类号:Q811.4 文献标识码:A 文章编号:1674-098X(2019)02(b)-0136-02
当前生物信息测序领域中,Illumina公司属于二代测序的垄断公司。其开发的Illumina Hiseq、Illumina Miseq、Illumina GAII等平台已经是流行于全世界。绝大多数分子生物学、基因组学和细胞生物学实验室都在使用Illumina平台测序。
在二代测序的分析流程中,拿到的数据是FASTQ数据,需要先对数据进行质量控制。质量控制通常是使用FastQC软件对FASTQ数据进行分析,判断测序数据是否具有高质量。如果质量低,则不支持后续生物信息学分析,需要过滤。通常情况下,由于测序仪机器的误差,从测序仪下机的数据都或多或少有低质量的序列,这些低质量需要有的是碱基质量低,有的是测序接头未去掉(尽管Illumina大多数测序平台的测序仪在2013年以后能保证数据下机自动去接头,但是部分测序平台依然不能自动去接头)。需要过滤,而过滤使用的软件一般为Trimmomatic软件。但是Trimmomatic软件过滤使用的文件是自身adapter文件夹中自带的Truseq文件过滤测序的接头,而这些接头文件只包含了Hiseq、Miseq和GAII测序平台的接头文件,没有包括全部的接头文件。未去接头的序列在质控结果中可以将接头序列以Overrepresent形式表示出来。所以,如果我们测序时选择的测序平台不能自动下机去接头,我们需要手动自己去接头。爬虫是利用计算机技术对网络数据的挖掘,因为互联网数据基本都是储存在网络服务器中,网络服务器末端端口是用户。用户可以通过网页访问网络服务器,网页是由HTML语言搭建的可视化端口。HTML是HyperText Markup Language(超文本标记语言)的缩写,这个语言使用<标签>内容基本格式进行网页编辑[1]。例如
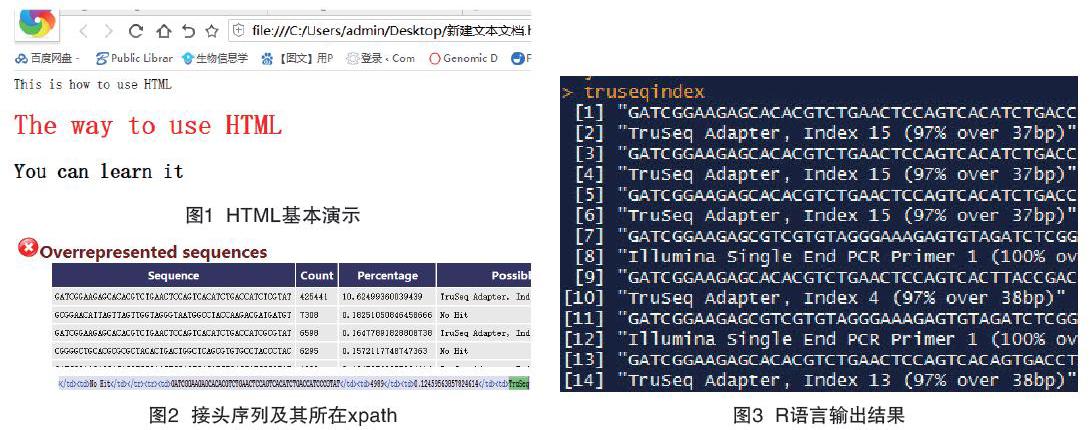
This is how to use HTMLThe way to use HTML
You can learn it
将上面这个代码复制到一个新建TXT文本中,并将后缀命名为.html,双击该文件打开会出现以下内容(见图1)。
1 分析方法
FASTQC软件输出的质控结果就有HTML本地文件,如果出现了接头序列就会在Overrepresent中出现,Overrepresent有其对应的HTML标签。R语言[2-3]可以通过追溯内容所在的标签追溯到内容,这个追溯内容的路径称之为xpath,最后通过正则表达式筛选我们要的内容即可。首先在Linux系统上存放测序数据的路径(该路径中只能含有测序数据文件)下使用命令fastqc `ls $pwd`,然后下载输出的HTML文件。先用网页查看是否有接头序列,再用Notepad++打开文件找接头序列所在的xpath(见图2)。
编写如下R语言代码
library(rvest)
library(stringi)
setwd("D:/test/fastQC")
myQCfile<-dir("D:/test/fastQC")
truseqindex<-NULL
for (i in 1:length(myQCfile)) {
qc<-read_html(myQCfile[i])
a<-qc%>%
html_nodes(xpath = "//tr/td")%>%
html_text()
b<-NULL
for (j in 1:length(a)) {
if(grepl(a[j],pattern = "(TruSeq|Primer)")){b<-c(b,a[j-3],a[j])} }
truseqindex<-c(truseqindex,b)}
2 结语
我们开发的挖掘当前过滤软件无法过滤的接头脚本更具有实用性,可以适用于所有NGS数据分析过滤脚本。使分析结果更具有可靠性。
参考文献
[1] 邓子云.爬虫系统中标签删除功能的设计及优化[J].计算机系统应用,2019,28(1):176-181.
[2] 许庆炜.B语言—生物信息学可视化流程语言[J].计算机与数字工程,2009,37(5):90-93.
[3] 吴栋杨. 构建基于R语言的生物信息学研究平台[A].第十次中国生物物理学术大会论文摘要集[C].中国生物物理学会,2006:1.
猜你喜欢
房地产导刊(2022年10期)2022-10-18
现代信息科技(2021年21期)2021-05-07
电子测试(2018年1期)2018-04-18
电子制作(2018年2期)2018-04-18
电子制作(2017年9期)2017-04-17
中国校外教育(下旬)(2016年11期)2016-12-27
中国教育信息化·基础教育(2016年10期)2016-12-20
今传媒(2016年11期)2016-12-19
电子设计工程(2015年6期)2015-02-27
