
采用高通量测序研究足癣患者真菌群落的种类
2019-06-10 10:30:24宋鹏飞陈加媛
中国麻风皮肤病杂志 2019年6期
贺 勤 宋鹏飞 陈加媛 徐 娟 王 京
足癣在全世界广为流行,在热带和亚热带地区更为普遍。在我国足癣的发病率也相当高。基于传统的培养方式研究发现,主要的致病菌种以皮肤癣菌为主,但是念珠菌等其他致病菌的感染也有明显增加[1]。尽管各种新的培养技术不断出现,但传统方法费时费力,灵敏度和特异性也较低,不能反应全部的真菌群落信息,这很大程度上影响了足癣的诊断和治疗。
与传统的培养方法相比较,耗时短、通量大、准确性高的分子生物学方法越来越普遍的用于分析和鉴定真菌群落多样性。近年来,高通量测序技术已经开始影响真菌生物学,逐步成为研究真菌群落多样性的一个新的发展方向和热点,增加了获得新发现的机会[2,3]。本文采用高通量测序的方法,针对足癣患者足部真菌群落进行分析和比较。通过大数据的挖掘,了解足癣患者足部真菌群落结构、真菌的种类和数量分布特点。为进一步优化和控制真菌群落结构提供参考数据,在预防和治疗中可根据具体的真菌感染对其进行各个击破,实现个体化用药和精确医疗的需求。
1 资料和方法
1.1 研究对象 收集我院门诊就诊的足癣患者,具体入组标准为:(a)临床诊断为足癣,镜检或培养结果呈阳性;(b)3个月之内未用过任何外用或口服抗生素类治疗药物。入组者均阅读并签署知情同意书。填写信息采集记录表(性别、年龄、病灶部位等信息)。
1.2 研究方法
1.2.1 样本采集 受试者的临床类型主要为浸渍糜烂型和丘疹鳞屑型,采集部位分别为受试者的趾间和足底。使用消毒的棉签拭子,生理盐水浸润后,在受试者真菌感染的皮损部位和对称健康部位反复刮取50次,获得局部真菌群落。样品采集后放入采样管,并立即送至-80℃冰箱保存。
1.2.2 总DNA提取及高通量测序 使用E.Z.N.A.® Soil DNA Kit(Omega Bio-tek, Norcross, GA, U.S.)试剂盒,按照试剂盒说明提取采集样品总DNA。选用特异性引物ITS1F(5’-CTTGGTCATTTAGAGGAAGTAA-3’)和 ITS2R(5’-GCTGCGTTCTTCATCGATGC-3’),以微生物总DNA为模板进行PCR扩增,每个样品引物加入8bp的标签序列,用于区分不同的样品,每个样品做3个重复。PCR扩增条件:95℃ 5min;95℃ 30s,55℃ 30s,72℃ 45s,27个循环;72℃ 10 min。2%琼脂糖凝胶电泳检测 PCR 产物后,采用AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, U.S.)回收纯化目的条带,采用QuantiFluor: trademark: ST(Promega, U.S.)对回收片段进行定量。回收片段按照定量混合后采用Illumina HiSeq 2500测序平台PE 250测序策略进行高通量测序。
1.2.3 高通量数据分析方法 过滤后的下机数据采用FLASH软件(V1.2.7,http://ccb.jhu.edu/software/FLASH/)拼接序列,使用Qiime软件(V1.9.0,http://qiime.org/index.html)进行序列质控。使用 Uparse软件(Uparse v7.0.1001,http://drive5.com/uparse/)对所有样品的全部有效序列进行97%水平聚类形成可操作分类单元(Operational Taxonomic Unit, OTU)。使用Usearch软件通过Silva(v128, http://www.arb-silva.de)进行物种注释(置信度阈值为0.8)。后续使用Qiime软件进行α多样性的计算分析。使用R语言Vegan包中的Anosim分析样品组间真菌群落结构的差异。应用方差分解,对多维数据降维,进行主成分分析(Principal Component Analysis, PCA),从而提取出数据中最主要元素和结构。使用Metastat软件进行组间差异物种统计分析。
2 结果
2.1 有效样品的数据统计 成功获取15例患者的24个有效样品。病灶组(D组)15个样本,其中取样在趾间的9个样本(DA组),取样在足底的6个样本(DB组);健康组(H组)9个样本,其中取样在趾间的4个样本(HA组),取样在足底的5个样本(HB组)。24个样品通过Illumina平台HiSeq2500测序获得的数据进行低质量数据过滤、拼接、去除嵌合体序列后,获得的拼接序列平均长度在190-300bp之间,每个样品有效序列数目在3万条以上。经过97%相似性聚类之后,剔除未注释到的序列,共计获得了1061个OTU。
2.2 真菌物种注释分类结果 对全部OTU进行物种注释,共检出6个菌门,分别为:担子菌门(Basidiomycota)、子囊菌门(Ascomycota)、接合菌门(Zygomycota)、壶菌门(Chytridiomycota)、球囊菌门(Glomeromycota)和罗兹菌门(Rozellomycota)。其中子囊菌门和担子菌门为主要优势菌门,所占丰度比例分别为61.18%以及38.04%。两个组共检出283个属,其中丰度最高的10个属分别为毛癣菌属(Trichophyton)、毛孢子菌属(Trichosporon)、曲霉属(Aspergillus)、镰刀菌属(Fusarium)、念珠菌属(Candida)、红酵母属(Rhodotorula)、枝孢属(Cladosporium)、皮毛孢子菌属(Cutaneotrichosporon)、枝孢霉菌属(Amorphotheca)、青霉属(Penicillium)。毛癣菌作为丰度最高的真菌,是足癣的主要致病菌,在D组中的丰度(24.98%)显著高于H组(6.22%)。见表1。
2.3 组间真菌多样性分析 α多样性分析是对样品中物种多样性的分析,包含样品中的物种组成的丰富度和均匀度两个因素,通常用Shannon,Chao1,Observed species以及PD Whole Tree等指数来评估样本的物种多样性,指数越高,表明样本的多样性越复杂。对两组样品进行α多样性分析,结果如图1所示。结果显示两组样品在这4种物种多样性水平上差异均不显著(P值分别为0.305,0.0721,0.0725,0.224),说明足癣患者病灶部位的真菌群落多样性与对称健康部位的真菌群落多样性相近。

表1 足癣患者足部真菌种类丰度 (%)

图1组间真菌多样性比较
2.4 组间真菌群落结构的差异 根据取材部位不同,又将病灶组和健康组分成了趾间病灶组(DA)、足底病灶组(DB)、趾间健康组(HA)和足底健康组(HB)。Anosim分析显示:DA与HA两组间真菌群落结构差异显著,其它三组间差异不显著。见表2。

表2 组间真菌群落结构差异分析
对DA与HA两组样品进行PCA分析发现:DA与HA组样品均具有明显的聚集,DA组本身又分为偏裸囊菌科(Arthrodermataceae)以及偏毛孢子菌科(Trichosporonaceae)型两种类型,而健康对照HA组更倾向丛赤壳科(Nectriaceae)。将DA组按照PCA分布拆分分组为DA1和DA2两组,通过Metastat检验结论为毛孢子菌科在DA2组的丰度显著高于DA1组以及HA组(P=0.00,P=0.00);裸囊菌科在DA1组与DA2组的丰度显著高于HA组(P=0.00,P=0.00),见图2。基于各样品的在科水平上的物种丰度信息进行主成分分析,用分析得到的第1、2主成分对样品进行画图,得到图2a结果。其中PC1表示由样本相对丰度信息提取到的第1主成分,31.6%表示主成分1对样本科水平差异的解释率;PC2表示由样本丰度信息提取的第2主成分,20.9%表示主成分二对样本科水平差异的解释率。箭头连线与横竖坐标轴的夹角表示该物种与该主成分的相关性,夹角越小表示相关性越小。如,裸囊菌科与PC1的夹角大于与PC2的夹角,说明裸囊菌科与第1个主成分的相关性比第2个主成分高。进一步分析发现,裸囊菌科中毛癣菌属为优势菌,毛孢子菌科中毛孢子菌属为优势菌,丛赤壳科中镰刀菌属为优势菌。
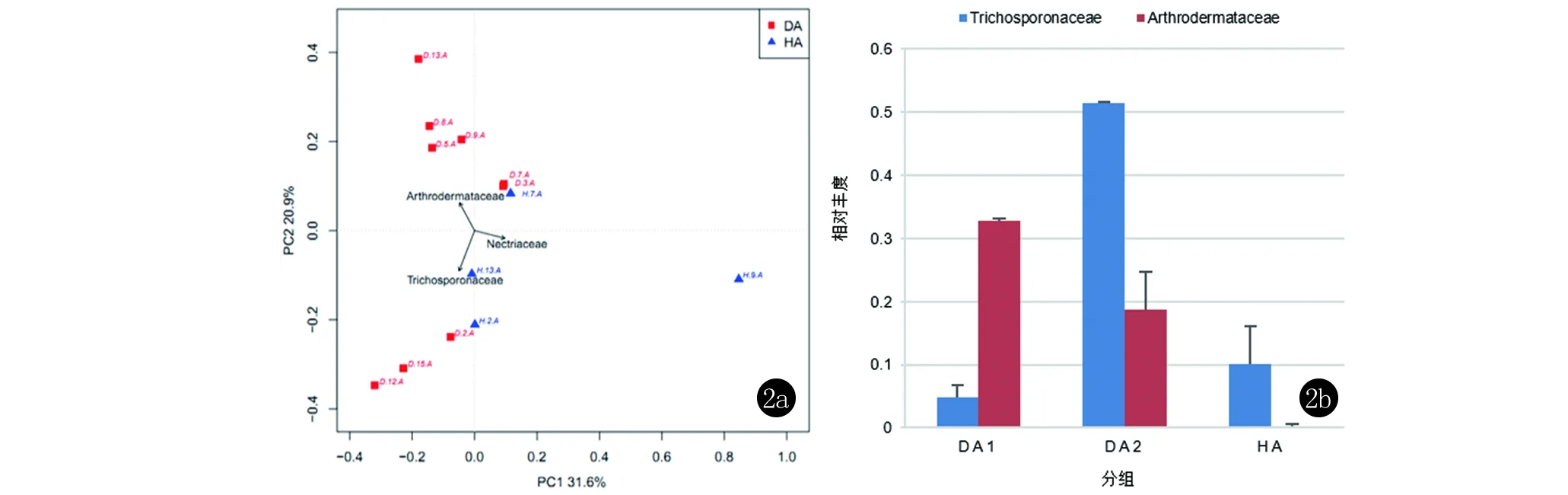
图22a,2b PCA分布(科水平)及相关物种丰度
3 讨论
皮肤是人体的最大器官,其上定植着大量的微生物菌群,在防御和对抗病原体入侵、免疫系统训练及稳态维持中起着重要的作用。寄生在皮肤表面的微生物群落与人体相互依存、相互影响,形成了在人体皮肤结构功能中发挥重要作用的微生态环境。在微生态系统被破坏的情况下,可能导致皮肤病甚至全身性疾病[4]。因此研究皮肤微生物群落的组成特点对于阐明皮肤病的病因学是有价值的。目前国内外对于银屑病、特应性皮炎的微生物菌群有较多的研究[5,6]。对于足癣这类病种的微生物群落研究,罕有报道。
本次研究中,足部皮肤真菌群落以子囊菌门和担子菌门为主要优势菌门,这一分布特点与以往的研究结果一致[7]。所有样本总共检测出283个属,说明足部真菌种类丰富,群落结构的不稳定性较高,可能为致病菌提供感染机会。病灶组的优势菌为毛癣菌属,它的含量显著高于健康组,符合毛癣菌是足癣主要致病菌的传统研究结果[8]。另外,毛孢子菌属、曲霉属、念珠菌属、红酵母属等这些条件致病菌也在足癣患者的病灶部位或健康部位定植分布,它们的存在是否影响局部皮肤的微生态稳定,是否参与机体的免疫调节,在足癣的发生发展过程中起到了何种作用,这些都有待于进一步研究和探讨。
对于真菌多样性分析的结果,我们发现足癣患者的病灶部位真菌群落多样性和对称的健康部位相近,说明多样性水平的高低对于足部健康的影响有限。但在我国北方的研究人员发现,足癣患者足部的真菌群落多样性要显著低于健康人群[9]。研究结果的不一致,可能与取材的具体部位、皮损的患病程度以及研究所处的地理位置和气候环境差异有关[2,7]。我们还需加大样本数量,强化分析方法,进一步验证和精确我们的分析结果。
本次研究我们还发现,足癣患者病灶趾间部位的真菌群落结构与对称的健康部位存在差异。根据优势菌种的分布不同,DA组又显著分成了DA1组和DA2组,分别富含致病菌毛癣菌属和条件致病菌毛孢子菌属,说明不同足癣患者趾间部位的优势菌种有所不同,这可能是足癣进行单一治疗方案效果不佳的原因。对于足癣患者在今后的诊疗过程中,应针对真菌群落组成,采用个性化治疗方案。
猜你喜欢
农家致富顾问·上半月(2022年11期)2022-05-30 03:56:07
武警医学(2022年3期)2022-04-07 06:43:56
昆明医科大学学报(2022年2期)2022-03-29 00:51:58
食品安全导刊(2021年20期)2021-08-30 06:40:50
中国民间疗法(2021年1期)2021-04-20 02:31:00
饲料博览(2019年8期)2019-02-13 16:46:10
中国工作犬业(2017年5期)2017-06-06 02:06:38
农业知识(2016年15期)2016-05-25 09:45:04
水生生物学报(2015年1期)2015-02-28 16:01:05
中华皮肤科杂志(2014年4期)2014-12-19 12:55:43
