
网纹蟒非入侵式采样和线粒体基因组分析
2019-06-04 02:17何叶艳高旭龙赵阿勇
浙江农林大学学报 2019年3期
白 天,何叶艳,高旭龙,赵阿勇,何 珂
(浙江农林大学 动物科技学院,浙江 杭州 311300)
网纹蟒Python reticulatus属于蛇亚目Serpentes原蛇附目Henophidia蟒科Pythonidae,广泛分布于东南亚,在印度尼西亚存在海岛变体[1],其皮张被认为是皮毛产业奢侈品的来源,是东南亚重要的经济自然资源。目前,该物种被世界自然保护联盟评估为易危等级(vulnerable species,VU)[2],属于濒危动植物种国际贸易公约附录Ⅱ物种。受捕猎影响,该物种在东南亚各国不同地区的种群遗传多样性已遭到破坏,亟待有计划的种群复苏和保护工作[3]。蛇类进化历史久远,生存环境复杂多样,但其形态特征相对较为保守,加上趋同进化,造成可用于蛇类系统学研究的形态方面信息较少,因此,依据形态学信息很难解决蛇类系统关系。生物分子所具有的信息量大、完全不相同、较为真实记录生物进化信息等特点,可弥补形态学信息的不足,越来越多地被应用到蛇类系统发生的研究中[4]。研究认为:作为一个与能量代谢有关的细胞器,真核生物的线粒体具有相对独立的遗传物质即线粒体DNA(mitochondrial DNA,mtDNA),研究线粒体基因组并利用线粒体DNA全序列分析进行系统建树,能够克服单个基因仅能带来的有限信息,更加全面地揭示物种进化与系统发生。目前关于网纹蟒的分子生物学研究仅集中在几个线粒体基因相关的系统分析上[3],在线粒体全基因组方面上存在欠缺。本研究拟采用无损伤性取样,提取网纹蟒线粒体DNA,通过聚合酶链式反应(PCR)扩增拼接得到线粒体基因组全序列,以评估非入侵式采样在蛇类研究中的可行性;以网纹蟒线粒体基因组序列信息和结构特征分析为依据,结合GenBank中近缘物种(蟒科,蚺科Boidae和闪鳞蛇科Xenopeltidae)已发现的线粒体基因组序列,构建系统发生树,探讨蛇亚目的系统发生关系,并结合蛇亚目线粒体基因组不同的排列顺序来讨论蛇亚目线粒体基因组的重排。
1 材料与方法
1.1 试验样品
试验所用网纹蟒的蛇蜕和新鲜粪便均来源于杭州动物园,于每日喂食时观察并收集得到。粪便用体积分数70%乙醇浸泡,蛇蜕用体积分数95%的乙醇固定,带回实验室后保存于-20℃的冰箱中备用。
1.2 基因组DNA的提取
粪便样品分剥离表面和内层部分2种,蛇蜕样品剪碎后一部分室温下研磨,另一部液氮研磨至粉碎。各样品取3次重复,基因组DNA提取参考文献[5]方法。经NanoDropTM3300检测计算质量浓度和纯度D(260)/D(280)。
1.3 片段扩增
根据亲缘物种的线粒体基因组序列设计扩增引物,采用高保真酶(PhantaSuper Fidelity DNA Polymerase,诺唯赞,南京)进行扩增,PCR产物经电泳确定大小符合要求时,交由上海博尚生物公司测序。若PCR产物呈现多条带,则采用割胶回收试剂盒获取目的大小片段,用pMD-19T质粒(TaKaRa,大连)连接,选取阳性克隆后送出测序。
1.4 数据分析
(1)网纹蟒线粒体基因组各基因、tRNA长度和基因起始密码子分析。用Dnastar软件包拼接测序序列, 用 tRNA scan(http://lowelab.ucsc.edu/tRNAscan-SE/)确定 tRNA 的位置和类型, 采用 OGDRAW(https://chlorobox.mpimp-golm.mpg.de/OGDraw.html)绘制网纹蟒线粒体结构及基因结构图。在分析过程中,网纹蟒记为 RP,以蟒科的缅甸蟒Python bivittaus(PB,NC_021479),印度蟒Python molurus(PM,NC_015812)和球蟒Python regius(PR, NC_007399)作为对比。 (2)根据相关基因、 控制区(control region)和线粒体基因组全序列建树分析蟒科、蚺科和闪鳞蛇科的进化关系。用Mega 5.0软件[6]比对序列及构建系统进化树,蟒科物种为缅甸蟒、印度蟒和球蟒,蚺科物种为红尾蚺Boa constrivtor(BC,NC_007398)和玫瑰蚺Charina trivirgat a(CT, GQ200595), 闪鳞蛇科物种为闪鳞蛇Xenpeltis unicolo r(XU,NC_007402),以原蛇亚目的圆环蛇Anilius scytale(AS,GQ200593)作为外类群。(3)蛇类具有多种线粒体结构排布次序,采用OGDRAW软件绘制各蛇类线粒体基因结构图可清楚地分析线粒体基因在进化过程中的变动。对分属不同科属的11个物种构建系统进化树,分别为缅甸蟒、球蟒、红尾蚺、闪鳞蛇和存在1个控制区和tRNA-WQANCY基因簇的细盲蛇科Leptotyphlopidae物种西南细盲蛇Rena humilis(RH,NC_005961),含有2个控制区和存在tRNA-P复制的蝰科Viperidae物种冲绳烙铁头Ovophis okinavensis(OO,NC_007397)和美洲蝮Agkistrodon piscivorus(AP,EF669477),含有2个控制区和存在tRNA-P复制的游蛇亚科Colubridae物种北美玉米蛇Pantherophis slowinskii(PS,NC_009769)和半棱鳞链蛇Dinodon semicarinatus(DS,NC_001945),rRNA 长度存在增加和含有 tRNA-L(Q)M和 tRNA-W(ANCY)结构的疣鳞蛇科Acrochordidae物种疣鳞蛇Acrochordus granulatus(AG,NC_007400)和管蛇科Cylindrophiidae物种红尾管蛇Cylindrophis ruffus(CR, NC_007401)。
2 结果与分析
2.1 样品基因组检测
对提取到的DNA的检测发现,蛇蜕样品DNA质量浓度最高,粪便表层次之,粪便内层最低;粪便内层样品D(260)/D(280)最高, 说明样品 DNA 纯度较差, 其他样品的D(260)/D(280)均为 1.7~1.8, 符合后续实验要求(表1)。PCR扩增试验显示:室温或液氮研磨,蛇蜕样品中提取的基因组DNA均能扩增出长度为1 000~1 500 bp的片段;而来源于粪便表层或内层样品的基因组扩增得到700~1 000 bp片段和1 000~1 500 bp片段的效果均不佳。
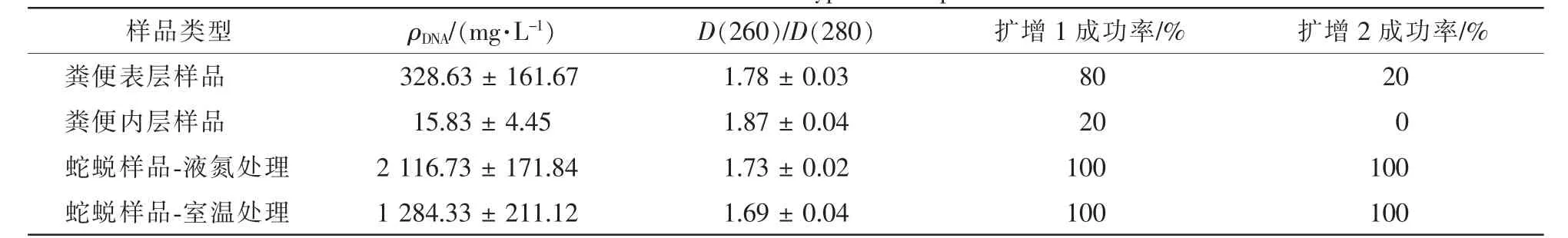
表1 所采用的样品类型提取DNA和扩增情况Table 1 Genomic DNA extracted from different types of samples and the success rate of PCR
2.2 蟒科蛇类线粒体基因组分析
对PCR扩增产物进行测序和序列拼接,得到长度为17 641 bp的网纹蟒线粒体基因组完整序列(GenBank:MH410033)(图1),序列含2个rRNA基因,13个编码区,22个tRNA基因和2个控制区(表2)。其中重链编码28个基因,轻链编码9个基因。包含的2个rRNA基因(12S rRNA和16S rRNA)位置和其余蛇类相同。除了NADH6(ND6)之外,其余12个蛋白编码基因均位于重链上;所有蛋白基因都没有内含子,但和相邻的基因存在少许重叠或间隔。发现的22个tRNA基因存在蛇类特有的LQM和WANCY基因簇。2个控制区位于非编码区,长度分别为1 271和1 196 bp,序列相似度为96.68%。
对蟒科物种相关基因的长度、起始密码子和终止密码子的分析发现,4个物种之间存在较高的同源性。网纹蟒的NADH2(ND2)终止密码子和ATP8起始密码子与其他物种不同,是物种所特有的;NADH6(ND6)基因变异最大,起始密码子仅2个物种一致,终止密码子缅甸蟒不同于其余3个物种。COX2基因和NADH1(ND1)较为保守,前者4个物种均以TA为终止密码子,后者则使用单碱基T作为不完全终止密码子。
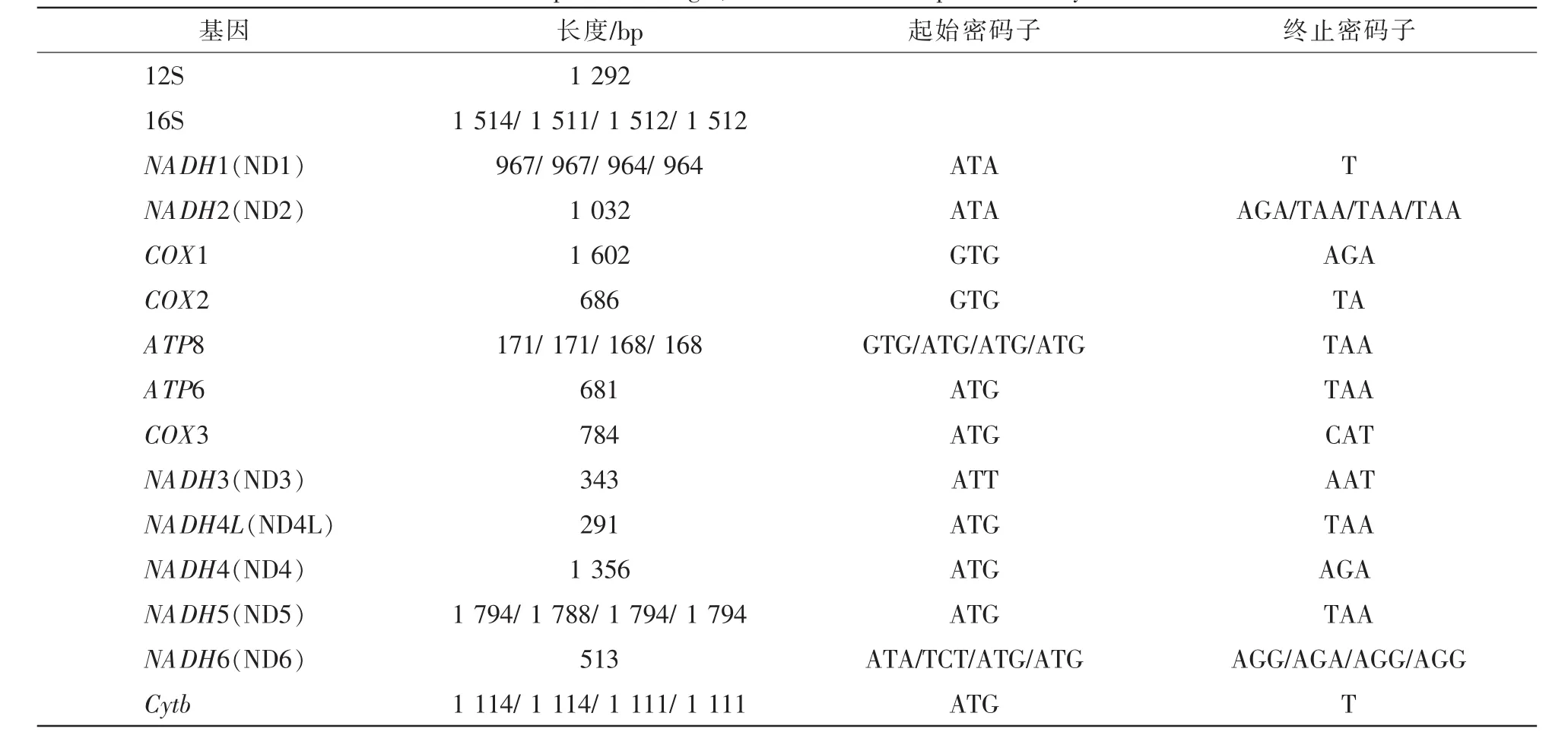
表2 蟒科蛇类的线粒体蛋白质基因长度、起始密码子、终止密码子和rRNATable 2 Comparion of length,start codon and stop codon in Pythonidae
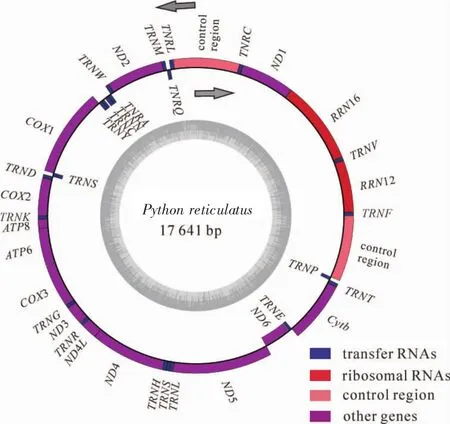
图1 网纹蟒线粒体基因组结构示意图图1 Schematic diagram of mitochondrial genome structure in Python reticulatus
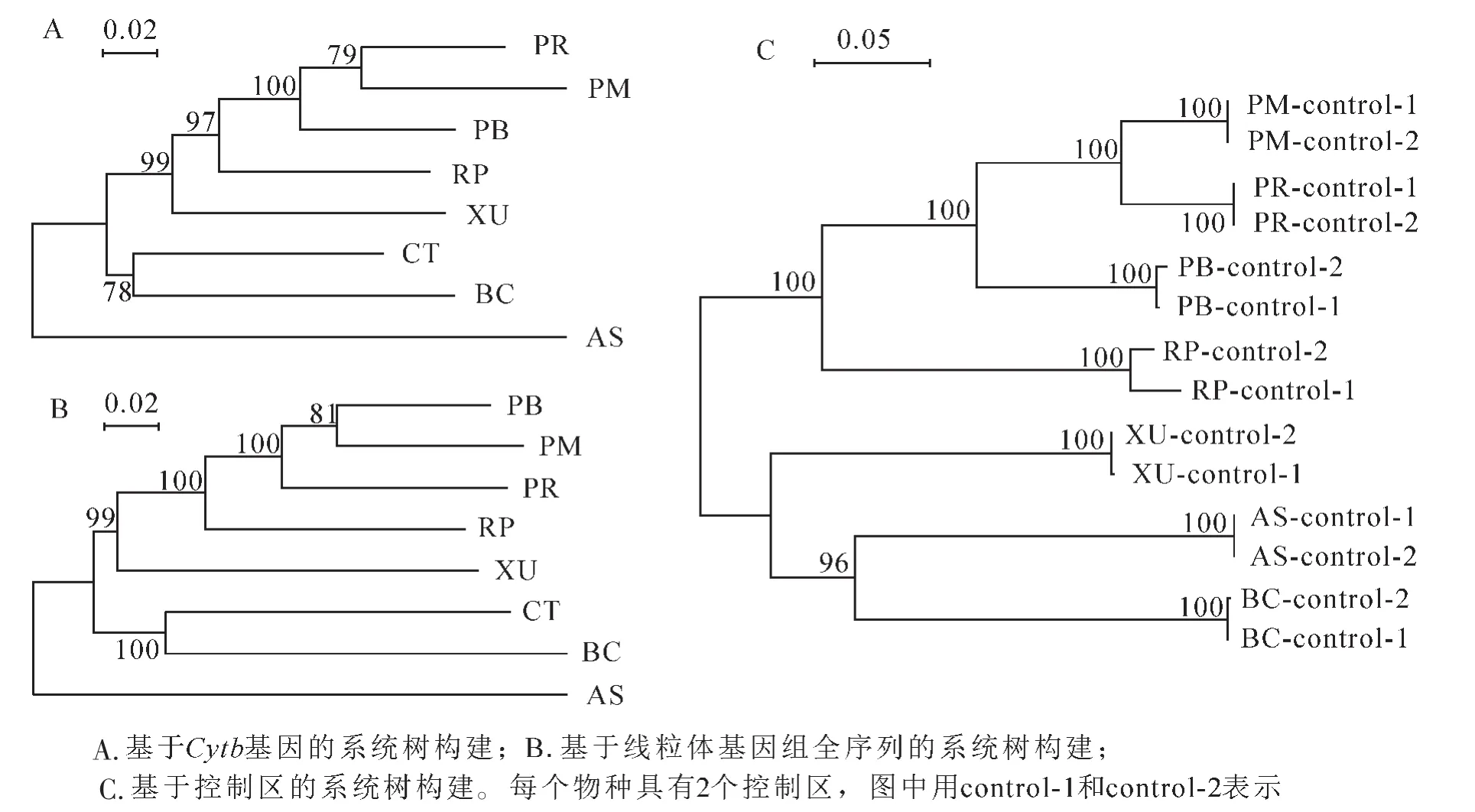
图2 蟒科、闪鳞蛇科和蚺科的系统发育树Figure 2 Phylogenetic relationship between Pythonidae,Xenopeltidae and Boidae
2.3 蟒科和蚺科物种进化分析
利用相关基因、控制区和线粒体基因组全序列构建系统树。分析发现:用蛋白基因构建的系统树拓扑结构较为相近;以Cytb基因为例(图2A),蟒科物种聚集的支持率为97%(PR,PM,PB和RP枝),就亲缘关系而言,闪鳞蛇科(XU)比蚺科(BC和CT)更接近蟒科(图2A)。用线粒体基因组全序列构建的系统树则表明:蟒科(PR,PM,PB和RP枝)和蚺科(BC和CT枝)各自聚为一枝,支持率都为100%;同样,闪鳞蛇科比蚺科更接近蟒科(支持率99%)(图2B)。由于数据库中玫瑰蚺(CT)的2个控制区长度仅为288和553 bp,和其他数据存在明显差异,因此删除该序列后对其他7个物种进行基于控制区的系统树构建。分析发现:各物种的2个独立的控制区均先聚类,物种间的控制区聚类关系表明蟒科的支持率为100%,而闪鳞蛇、蚺科和圆环蛇则为另外一分支(图2C)。以上结果表明:来源于同个物种的控制区相似度更高;相较于蚺科,闪鳞蛇科和蟒科的进化关系更接近。
2.4 蛇亚目物种线粒体结构排步分析
和其他脊椎动物线粒体基因组全序列的比较发现,蛇亚目物种长度大小变化较大,主要原因为控制区的长度和数量不同,基因组上存在一些重复序列区域。对不同物种线粒体结构序列进行分析并构建系统树(图3),结果发现存在3种不同的基因结构,结构一主要存在于蟒科(RP和PR),闪鳞蛇科(XU),蚺科(BC)和疣鳞蛇科(AG 和 CR)中,含有 2 个控制区及 tRNA-L(Q)M和 tRNA-W(ANCY)结构; 结构二存在于蝮亚科(OO和AP)和游蛇科(DS和PS),和结构一的差异在于在tRNA-I和控制区1中多了1个tRNA-P;相比于结构二,结构三的tRNA-L(Q)M基因簇中,tRNA-Q产生了基因轻重链的转化,形成了tRNA-LQM;而结构四(代表为盲蛇总目RH)则为仅存在1个控制区,tRNA-L产生了移位,挪动至ND1前,而tRNA-L(Q)M和 tRNA-W(ANCY)通过移位分别转变为 tRNA-IM和 tRNA-W(QANCY)。
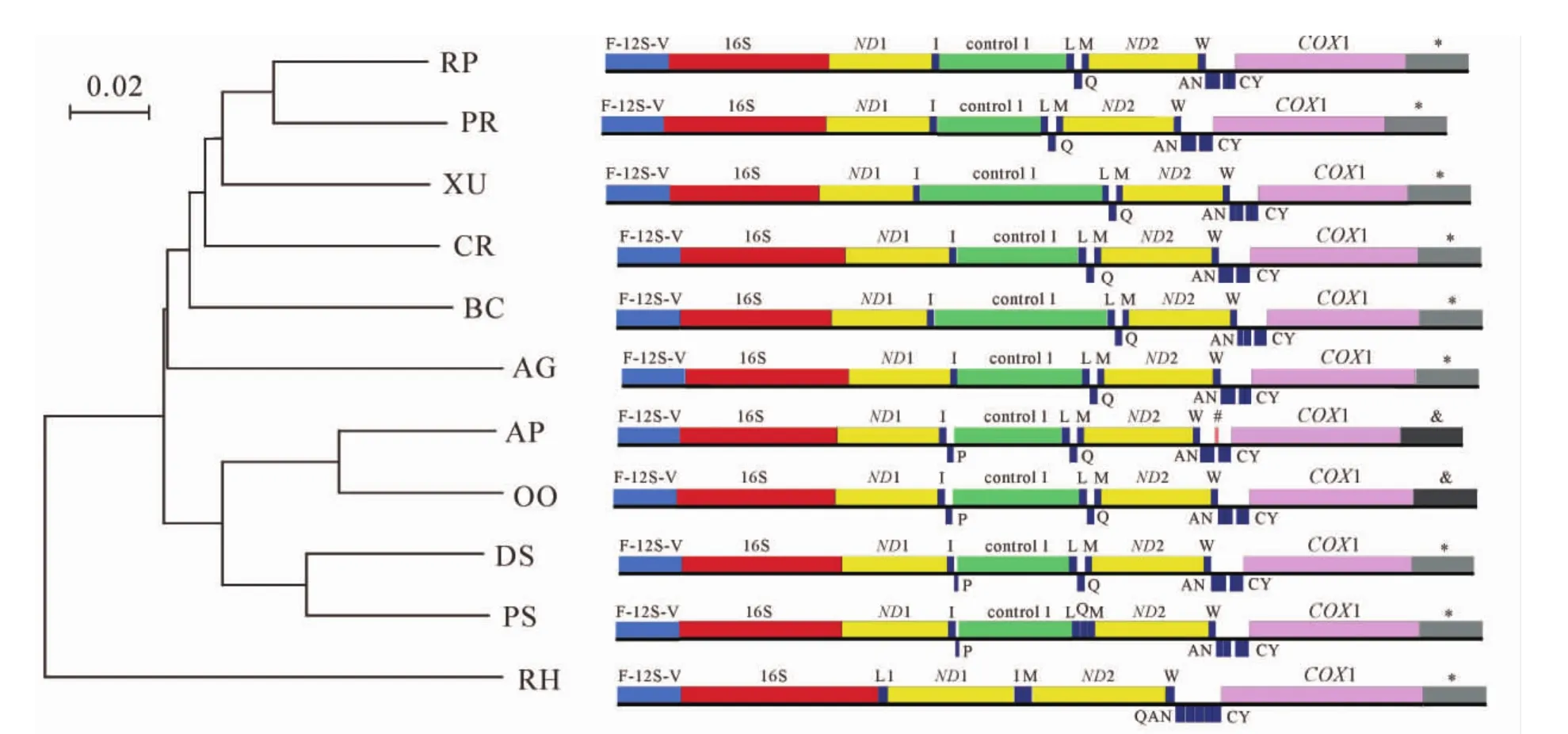
图3 不同基因结构的线粒体基因组系统进化树及基因结构示意图Figure 3 Phylogenetic tree of mitochondrion based on different gene structures and the rank of variable area
3 讨论
3.1 非入侵式采样在蛇类研究中的应用
在野生濒危动物中采集血液和脏器等常规样品非常困难,要建立濒危动物的物种基因库,亟待寻找新的采样方法。研究认为:通过提取粪便DNA或其他生物痕迹DNA等非入侵式采样方法能够为遗传学和生态学研究提供有效信息,在检测种群大小、物种饮食结构和大量物种的激素信息上具有现实意义[7]。蛇类的蜕皮行为为非入侵式采样提供了实例。KHEDKAR等[8]提取92份蛇蜕样品基因组,通过DNA条形码技术鉴定出了23种蛇类,但部分样品不能有效进行扩增。石林春等[9]在利用蛇蜕鉴定中药材中的蛇类物种来源时发现,采用线粒体细胞色素C氧化酶I亚基(COI)作为引物,扩增片段不超过1 000 bp。网纹蟒是目前已知最长的蟒蛇,在东南亚国家捕杀严重,已被列为易危等级。本研究以粪便样品和新鲜蛇蜕为材料提取基因组DNA,发现相较粪便样品,蛇蜕样品扩增得到的片段长度更长(1 000~1 500 bp),液氮研磨能有效提高DNA的提取品质,满足后续实验要求。粪便样品由于提取的DNA品质和质量浓度不佳,在长片段扩增中效率低下,因此不建议使用。
3.2 蟒科和蚺科物种进化
本研究第1次完整地解析了网纹蟒的线粒体全基因组结构,填补了网纹蟒线粒体基因组全序列的空白。分析发现:网纹蟒的基因结构、tRNA顺序和控制区位置等均与蟒科物种一致,但也发现存在物种特异的特征,如NADH2以AGA作为终止密码子,ATP8以GTG作为起始密码子,NADH6以ATA作为起始密码子,这3点均不同于其他蟒科已报道的物种(表2)。
关于蟒科和蚺科物种的分子系统相关研究,主要采用部分线粒体基因结合核基因进行;由于线粒体全基因不能通过简单扩增得到,在分析物种进化过程中尚未广泛使用[4,10]。但线粒体全基因组包含的信息更多,在解释进化关系时更具有说服力[11]。本研究结合多个线粒体基因(Cytb,12S和16S)和多个核基因的序列,分析蟒科和蚺科物种的进化关系,认为蚺科在这3个物种中先分化出来,而蟒科和闪鳞蛇科始终聚成姐妹群关系。单基因和线粒体基因组的系统进化分析同样得到上述研究结果 (图2A,图2B),进一步确定了蟒科、闪鳞蛇科和蚺科三者之间的进化关系[10]。
3.3 蛇亚目线粒体基因组的重排
线粒体基因排列次序在进化过程中较为稳定,但在两栖类、鱼类和一些有袋类等类群中发现存在基因重复、基因重排和基因缺失等现象[11-12]。蛇类线粒体基因组在演化过程中出现了某些基因排列顺序的变化,如OL缺失、tRNA-LQM基因簇和tRNA-WANCY基因簇中的基因重排、控制区重复、tRNA-L移位和tRNA-P的功能缺失等[11],进化程度高级的新蛇附目Caenophidia中重排更加多样[13]。由此认为:具有共同基因重排顺序的物种很有可能具有共同的祖先,因为不同物种间共享的基因排列顺序,不可能是通过趋同进化而独立产生的。
蟒科在分类上属于蛇亚目原蛇附目。结合基因结构和顺序分析,蟒科目前已报道的3个物种和本研究所报道的网纹蟒线粒体结构均体现出真蛇类物种的线粒体基因结构类型,存在tRNA-LQM基因簇和tRNA-WANCY基因簇,并且没有发生tRNA-P的假基因化现象,存在2个控制区,且结构基本统一。tRNA-LQM基因簇由早期真蛇类的tRNALeu发生移位形成。本研究发现:除细盲蛇科物种(RH)外,所列举的物种均含有该基因;而tRNA-WQANCY基因簇由tRNAGln移位生成,仅存在于细盲蛇科(RH);真蛇类tRNAPro假基因的出现,则表现为控制区2附近的tRNAPro假基因化(OO和AP)和控制区1附近的tRNAPro缺失(RH)。双控制区在其他爬行动物[14]、 两栖类[13]和鸟类[15]中也有存在, 重复控制区认为可能是功能上(使得可以从多个位点启动复制和转录,提高效率)和进化选择上的优势,多次复制和插入形成并维持双控制区[12]。
对控制区的聚类分析发现:物种中独立存在的2个控制区在系统发生过程中并非按排列的位置成簇聚集,而是在各物种间聚成一枝(图3C),这和之前其他蛇类中描述的结果保持一致[12]。控制区重复事件认为发生在7 000万a前[12]:在原始类群蠕蛇附目Scolecophidia和细盲蛇科中仅存在1个控制区,随着蛇类演化,真蛇类线粒体中出现了控制区重复的事件,而在此基础上基因重排演化出tRNA的移位和转化。但聚类结果中2个控制区并没有按照2个分布进行聚类,主要原因则是因为在物种进化过程中发生了协同进化,导致同个物种的2个控制区形成了较高的同源性,相似度达100%[12]。
猜你喜欢
遗传(2022年9期)2022-10-10
科普童话·百科探秘(2022年2期)2022-05-24
科普童话·百科探秘(2022年1期)2022-05-24
现代企业文化·理论版(2020年16期)2020-08-31
今日农业(2020年14期)2020-08-14
生物学教学(2018年3期)2018-11-29
农村百事通(2018年6期)2018-03-29
科技视界(2016年14期)2016-06-08
内燃机与配件(2014年4期)2014-04-19
湖北农业科学(2011年15期)2011-11-18
