
Be,Mg,Mn掺杂CuInO2形成能的第一性原理研究*
2019-06-04 05:31莫曼曾纪术何浩张喨杜龙方志杰
物理学报 2019年10期
莫曼 曾纪术 何浩 张喨 杜龙 方志杰†
1)(广西科技大学理学院,柳州 545006)
2)(广西科技大学材料科学与工程研究中心,柳州 545006)
1 引 言
透明导电氧化物TCO (transparent conductive oxide)不仅具有良好的电学性能,也具有良好的光学透射性能.人们将透明导电氧化物广泛应用在太阳能电池、平面显示、特殊功能窗口涂层等光电器件领域[1].常见的透明导电氧化物TCO薄膜如ZnO,ln2O3,SnO2等[2-4]作为新一类性能优越、用途广泛的TCO薄膜材料体系,已经工业化并得到广泛使用.然而,目前广泛研究和应用的TCO材料大多数是N型导电材料.为了能够制备出透明二极管以及更多透明导电器件,需要研制性能优越、工艺稳定以及能够实用化的P型导电TCO材料.如果将P型TCO材料与N型TCO材料进行复合得到透明导电器件,将使得透明导电氧化物在应用方面得到很大的发展.然而与数量众多的N型TCO材料相比,P型TCO材料非常少,而且P型TCO的导电性能与N型TCO的导电性能相差很大,通常情况下P型TCO的电导率要比N型TCO的电导率低3—4个数量级.由于缺乏性能良好的P型TCO材料,目前仍无法实现具有良好性能的全透明的P-N结,因此,研制性能优越、工艺稳定的P型TCO材料成为拓展透明导电氧化物应用所必须面对的课题.
1997年,日本的科学家首次设计出具有较高载流子浓度和高电导率的P型TCO材料—CuAlO2[5],这一研究成果发表在《Nature》上.CuAlO2具有铜铁矿结构,其室温电导率为0.95 S·cm—1,Seebeck系数为+138 (正值表明材料具有P型导电特性),光学禁带宽度为3.5 eV,在500 nm厚度下,CuAlO2的可见光透射率达到80%,表明该体系可以满足透明电子学的要求.虽然CuAlO2的电导率与ZnO等N型TCO材料的电导率相比仍比较低,但是这一具有里程碑意义的研究发现还是极大地吸引了人们对P型TCO材料的兴趣[6].在发现CuAlO2具有P型导电性能之后,一系列P型铜铁矿类TCO材料也陆续被发现具有P型导电特性[7-14].
在目前P型铜铁矿类TCO材料中,CuInO2作为具有极大带隙值的P型TCO材料,而且具有双性掺杂的性能,即可进行N型和P型掺杂.因此,CuInO2受到许多人的重视.但是众多实验结果[15-17]表明CuInO2材料高的电阻率和低的透光率仍旧是制约其发展的两大难题,如何进一步提高CuInO2材料的导电性能并同时保证其较高的光学性能是以后努力研究的方向之一,对CuInO2掺杂将成为解决这一问题的有效途径.而选用何种类型的掺杂元素、采用何种掺杂技术则是制备高质量和高发光性能铟基光电材料的关键所在.目前对CuInO2掺杂改性的研究已有报道[18-20],其中吴平等[21]用Sn,Ca元素对CuInO2材料里的In元素进行替位掺杂研究,发现Sn,Ca掺杂之后的杂质能级都是属于深能级,因此对提高CuInO2材料的导电性帮助不大.到目前为止,对CuInO2材料的掺杂元素选择主要为Sn,Ca元素,而对具有其他的掺杂金属则还没有涉及.为了更好地了解对CuInO2材料的掺杂性能,本文选择掺杂元素Be,Mg,Mn对CuInO2进行掺杂改性研究,分别对替代Cu、替代In、进入间隙位三种掺杂模式进行基于密度泛函的第一性原理计算,研究掺杂元素在CuInO2材料中的微观作用机理,研究成果可为设计和开发高效率的新型光电材料提供崭新的研究思路.
2 计算方法和计算模型
CuInO2的晶体结构属于R3m空间群,晶胞中具有3个特征结构单元:1)平行C轴分布的OCu-O哑铃结构;2)垂直C轴的六角Cu层;3)InO6八面体结构,其中In位于八面体内(见图1).总能计算中原子赝势则采用 投影缀加波(PAW)赝势[22,23],交换关联势采用局域密度近似(LDA)[24,25],采用软件包VASP进行计算[26,27].能带结构计算采用包含四个原子的原胞,在计算的超晶胞中则包括108个原子,计算过程中所有的原子都被弛豫到H-F受力最小.截断能设置为400 eV.布里渊区的K点设置为1×1×1 Monkhorst—Pack网格[28,29].对于带电荷的缺陷,计算中引入一个均匀电荷背景以保持整个周期性超晶胞的电中性,而且所有超晶胞计算都采用同样的K点设置.单质的化学势也是在LDA和PAW势的总能计算中获得.CuInO2材料中所考虑的价电子为Cu的3d和4p电子,O的2s和2p电子,In的5d和6s电子;对于掺杂元素Be,Mg,Mn,价电子包括Be的2s电子,Mg的3s和3p电子,Mn的3d和4s电子.

图1 CuInO2晶体结构图,图中红色原子为O原子,灰色原子为In原子,棕色原子为Cu原子 (a)黄色掺杂原子替代Cu原子的情况;(b)绿色掺杂原子替代In原子的情况Fig.1.The crystal structure of the CuInO2,the red atoms are O atoms,the brown atoms are Cu atoms,the purple atoms are In atoms:(a)Substituting yellow dopant atom for Cu atom;(b)substituting green dopant atom for In atom.
为了提高和调整CuInO2材料P型电导率,我们计算了掺杂形成能和缺陷跃迁能级.根据形成能的定义[30,31],形成能由单质原子的化学势和电子的费米能决定:

此处E(α,q)为有缺陷α并且带电荷q的超晶胞总能;E0(CuInO2)为没有任何缺陷的超晶胞总能;nCu,nO,nIn,nA分别表示在形成缺陷时组分Cu,O,In和掺杂元素A从超原胞中转移到原子池中的原子数;q为所带有的电荷数为标准化学势,可分别视为Cu (面心体)、In (面心体)、O (氧气)和掺杂元素的体系总能;EF为体系费米能,从价带顶VBM开始,取值范围为价带顶到导带底之间.在化学势的计算中,因为单质化学势是与实验中材料生长过程有关的,因此具有相应的范围.
对于CuInO2材料,组分中的化学势范围必须满足以下条件:
1)每种组分的化学势不能大于该元素单质构成的体材料化学势,即
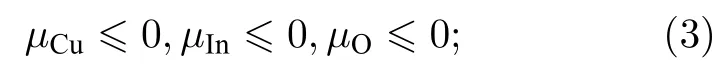
2)各组分的化学势相加为CuInO2,

CuInO2总能E0(CuInO2)= —651.22 eV,标准化学势μCu= —4.71 eV,μIn= —3.23 eV,μO= 5.24 eV,计算所得的CuInO2材料的形成能ΔHf(CuInO2)= —5.00 eV;
3)不能够产生组分之间的第二相,

4)掺杂的元素不能与原材料的元素组成第二相,即
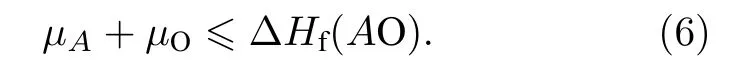
计算结果In2O3形成能ΔHf(In2O3)= —9.98 eV,Cu2O形成能ΔHf(Cu2O)= —1.67 eV.因此,CuInO2材料中的三种成分元素的化学势取值范围分别为:—0.84 ≤μCu≤ 0,—4.99 ≤μIn≤ —2.49,—1.67≤μO≤ 0.
3 计算结果与讨论
计算中,我们对CuInO2的晶格参数(a,c)和原子内部参数u进行了优化计算,CuInO2的晶格参数a和c是根据Murnaghan状态方程来优化拟合计算总能量,而原子内部参数u是根据原子的受力状况来进行优化,CuInO2晶格常数理论计算值与实验值[12]见表1.

表1 CuInO2晶格常数理论计算值与实验值Table 1.Theoretical values and experimental values of lattice constants in CuInO2..
掺杂元素的进入使得CuInO2晶体内部平衡状态受到了破坏,在掺杂元素周围的原子将发生不同程度的弛豫.表2列出了CuInO2中掺杂元素周围的弛豫变化情况,表中MgCu表示Mg原子以替位掺杂形式进入Cu原子位置,MgIn表示Mg原子以替位掺杂形式进入In原子位置,Mgi则表示Mg原子进入CuInO2间隙位置.键距后括号为掺杂原子的近邻原子个数和种类.从表2可以看出,与CuInO2的CuO键长1.85 Å(O,2),InO键2.18 Å(O,6)相比,Mg元素的替换并没有引起周围氧原子大的弛豫变化,而掺杂元素Be,Mn元素则引起了邻域原子较大的弛豫变化.
表3是掺杂下CuInO2在电中性状态下当q=0和µi=0 时的外在掺杂形成能 ΔE(α,q),由此得出在不同的化学势条件下缺陷形成能可以通过(1)式推导.由于掺杂形成能与化学势、费米能都有关系,可分为几种极值情况进行分析.在CuInO2材料中,化学势条件分两种情况,一种为有阴离子化学势极大(阳离子化学势极小)的情况,另外一种则为阴离子化学势极小(而阳离子化学势极大)的情况.
图2和图3分别为氧化学势极大和极小情况下,Mg,Mn,Be掺杂元素形成能随费米能的变化图,纵坐标表示形成能(形成能在零以下表示易于形成),实线表示掺杂的形成能随着费米能变化的过程,数字表示带电情况,小实心点表示跃迁能级,VBM为价带顶,CBM为导带底,费米能取值在VBM与CBM之间.当费米能EF移动接近导带底时,根据(1)式,受主类(Mg,Mn,Be替换In原子)的掺杂形成能就会降低,而施主类(Mg,Mn,Be替换Cu原子或Mg,Mn,Be进入间隙位)的掺杂形成能将会增高.由图2 (氧化学势极大)可以看出,氧化学势极大情况下,从掺杂元素的形成能分析,Mg,Mn原子都容易替换CuInO2里面的In原子,随着费米能级向导带底移动,Be原子也有替换In原子的可能;对Mg,Mn,Be替换Cu原子的情况,计算结果显示Mg,Mn,Be替换Cu原子的行为具有较高的形成能,表明这些掺杂元素很难替换Cu原子;同时也因为极高的形成能,Mg,Mn,Be掺杂元素进入CuInO2间隙位置的情况不会发生.而从图3可以看出,在氧化学势极小的情况,虽然Be,Mg都易于进入CuInO2,但是会同时形成受主类和施主类缺陷,这极大地影响了材料的导电性能和效率;随着费米能EF向导带底移动,MnIn这类形式的掺杂也能发生,而MnCu,Mni的形成能较大,难以形成在CuInO2材料中.

表2 掺杂元素周围的弛豫变化情况,键距后括号为掺杂原子的近邻原子种类和个数Table 2.The surrounding atoms of dopant,kinds and numbers of dopantxs nearest neighbor atoms in parentheses.

表3 CuInO2的掺杂元素形成能Table 3.The calculated formation energies of dopants in CuInO2.

图2 掺杂形成能在cation-poor,anion-rich的变化图Fig.2.The change of doping formation energies under cation-poor,anion-rich condition.

图3 掺杂形成能在cation-rich,anion-poor的变化图Fig.3.The change of doping formation energies under cation-rich,anion-poor condition.
图4为各种类型掺杂元素进入CuInO2后所形成的缺陷能级,在确定掺杂元素能级位置时,我们采用芯能级对齐方式[32]进行能级位置确定.从图4可看出,Ⅱ族元素Be,Mg替代In元素之后所形成的缺陷能级都很接近价带顶,MgIn和BeIn的位置分别为价带顶上0.05 eV和0.06 eV,均属于缺陷浅能级,而MnIn的位置位于价带顶上0.4 eV,属于深能级.这表明MgIn和BeIn很适合作为受主在CuInO2中产生空穴载流子,再结合图2和图3的形成能计算结果,Mg原子替换In原子在氧化学势极大的情况下能大量生成且不存在自补偿效应(MgCu和Mgi在该情况不会形成),因此在氧化学势极大的情况下,MgIn是CuInO2材料中最为理想的受主,并能较好地提高材料的P型导电性能.为了增加CuInO2中的P型导电性,通过原子层外延生长的控制,在MBE或MOCVD晶体生长过程中实现减少各类金属的偏压以及增加氧偏压的方法以增加Mg原子替代In原子的概率,这种选择性位置的掺杂方法能够提高材料的P型导电性能.而CuInO2的外在掺杂N型缺陷都具有较高的形成能或较高的跃迁能级,因此,对于Mg,Mn,Be元素,无论何种形式的掺杂均很难在CuInO2中实现N型导电性能.

图4 掺杂元素在CuInO2的缺陷跃迁能级Fig.4.The calculated transition energy levels of the extrinsic defects in CuInO2..
4 结 论
利用第一性原理计算方法,研究掺杂元素Mg,Be,Mn对CuInO2材料的作用机理,分别对替代Cu、替代In、进入间隙位三种掺杂模式进行计算.计算结果发现,Mg原子的替位掺杂不会引起CuInO2体材料内部原子弛豫,在氧化学势极大的情况下,MgIn能大量生成且缺陷位置在价带顶上方0.05 eV,同时也避免了Mgi,MgCu的形成,MgIn因此能成为CuInO2的理想受主缺陷,能较好地提高材料的P型导电性能.另外,因为较高的形成能和深的跃迁能级,Mg,Be,Mn三种元素替代Cu和进入间隙位的掺杂模式都不会给材料带来N型导电性能.
猜你喜欢
当代党员(2022年9期)2022-05-20
纺织科学研究(2021年7期)2021-08-14
中外文摘(2021年7期)2021-04-23
华人时刊(2021年23期)2021-03-08
发明与创新·小学生(2020年10期)2020-10-19
复旦学报(医学版)(2020年3期)2020-06-18
无机盐工业(2019年4期)2019-12-25
发明与创新·小学生(2019年12期)2019-12-05
中学生数理化·高三版(2017年1期)2017-04-20
科学与财富(2016年28期)2016-10-14
