
Ce/Co比对Ag/CeO2-Co3O4催化剂低温氧化降解甲醛性能的影响
2019-06-03 02:35:46卫广程王慧敏张秋林
燃料化学学报 2019年5期
唐 彤, 卫广程, 王慧敏, 刘 墨, 张秋林, 宁 平
(昆明理工大学 环境科学与工程学院, 云南 昆明 650500)
甲醛作为室内空气主要污染物之一,属挥发性有机化合物,主要来源于建筑和装饰材料的排放[1]。长期暴露其中会对人体造成严重的危害[2],去除甲醛改善室内空气质量具有重要的研究意义。
迄今为止,催化氧化法被认为是最有效的去除甲醛的技术,能够将甲醛完全氧化成CO2和H2O[3-6]。其中,贵金属催化剂因其优异的低温催化氧化性能而备受关注,负载型Pt[7,8]、Pd[9,10]、Au[11,12]催化剂能够在室温实现甲醛的完全氧化。然而,Pt、Pd和Au负载型催化剂因价格昂贵而难以广泛应用。目前,Ag基催化剂因其廉价及较好的低温活性受到研究学者的广泛关注。Bai等[13]将Ag浸渍到三维有序介孔MnO2上,所得Ag/MnO2催化剂具有较低的反应活化能,在110 ℃可实现甲醛的完全去除。Qu等[14]报道称当双金属Ag/Co质量比为3∶1时AgCo/APTES@MCM-41催化剂显示出最优活性,能够在90 ℃将甲醛完全氧化。为进一步降低Ag含量提高催化剂低温催化活性,Ma等[15]发现,通过加入碱金属Na,2.4%Ag-Na/CeO2在100 ℃显示出甲醛的完全转化。但低温催化性能仍需提高,且Ag的负载含量也仍需进一步降低。
过渡金属氧化物用于甲醛的催化氧化研究也被广泛报道,其自身不但可吸附并活化甲醛,而且能够在催化氧化过程中以价态变化的形式传递电子[16-18]。其中,Co3O4以其较高的低温催化氧化甲醛性能备受关注。研究通过介孔结构构建提高比表面积,优化表面氧物种,进而提高催化活性[5]。但单金属氧化物还原性能和低温活性有限,因此,过渡金属研究主要着眼于复合金属氧化物。CeO2因其优异的储氧-释氧能力,常被作为第二金属引入以提高氧化还原性能。已通过大量尝试改善催化剂的低温活性,尤其通过优化不同金属比例。Zhu等[19]通过在KMnO4与(NH4)2C2O4之间的氧化还原反应过程中,加入Ce(NO3)3制备了不同Ce/Mn比的Ce-MnO2催化剂用于甲醛的催化氧化,当Ce/Mn比为1∶10时,催化剂的活性最佳,100 ℃可实现甲醛的完全转化。Liu等[20]研究发现,通过调节3DOMAu/CeO2-Co3O4催化剂的Ce/Co比以优化表面元素价态提高低温催化甲醛的活性,约39 ℃便能够实现甲醛的完全转化。
目前报道的文献中,负载型Ag基催化剂中活性组分负载含量相对较高,且甲醛的完全转化温度也较高。基于此,本研究以过渡金属Co3O4和CeO2复合氧化物为载体,制备一系列低Ag含量的Ag/CeO2-Co3O4催化剂并将其应用于低温催化氧化降解甲醛。本研究主要考察不同Ce/Co比对催化剂活性的影响,并揭示造成不同甲醛降解效率的影响因素。此外,本研究通过原位漫反射红外光谱实验对催化剂表面甲醛降解过程中间体进行检测,推测其具体反应机理过程。
1 实验部分
1.1 催化剂的制备
采用共沉淀法制备CeO2-Co3O4载体。以六水硝酸铈(Ce(NO3)2·6H2O)和六水硝酸钴(Co(NO3)2·6H2O)为前驱体源,共称取20 mmol Ce(NO3)2·6H2O和Co(NO3)2·6H2O,将其按物质的量比Ce∶Co为1∶19、5∶15、10∶10、15∶5和19∶1分别溶解于去离子水中。用草酸作为沉淀剂,将适量1 mol/L草酸缓慢滴加至上述溶液中,(Ce+Co)/草酸物质的量比为1∶3。于室温剧烈搅拌1 h,静置2 h,过滤水洗数次,80 ℃干燥过夜,并在450 ℃焙烧4 h。制得载体分别标记为Ce1Co19、Ce5Co15、Ce10Co10、Ce15Co5、Ce19Co1。作为对比,另分别称取20 mmol Co(NO3)2·6H2O和Ce(NO3)2·6H2O按上述步骤制备纯CoOx和CeO2载体,标记为CoOx和CeO2。
采用过量浸渍法制备Ag/CeO2-Co3O4催化剂,称取1 g制得的CeO2-Co3O4或CoOx粉末于化学计量的AgNO3溶液中于60 ℃搅拌蒸干。所得固体80 ℃过夜干燥过后放入马弗炉内,350 ℃焙烧3 h,制得Ag的质量分数均为1%的Ag/CeO2-Co3O4和Ag/CoOx催化剂。所得样品粉末分别标记为Ag/Ce1Co19、Ag/Ce5Co15、Ag/Ce10Co10、Ag/Ce15Co5、Ag/Ce19Co1、Ag/Co。Ag的理论负载量为1%。
1.2 催化剂的表征
X射线衍射谱图(XRD)由德国布鲁克生产的Bruker D8 Advance测试,扫描速率6(°)/min,10°-80°扫描。主要参数为Cu靶,管电压40 kV,管电流40 MA,步长0.02°。
氮气吸附-脱附实验用美国Micromeritics公司的TriStarⅡ3020型物理吸附仪测定样品的比表面积、孔容和平均孔径。样品在200 ℃下真空预处理3 h后,在-196 ℃下,以N2为吸附质进行测量。
拉曼光谱(Raman)测试由Renishaw inVia Reflex 2000型激光显微拉曼光谱仪完成。显微镜配备有Leica显微镜系统和50×物镜,使用Ar激发光源(λ= 532 nm)。在进行实验之前,拉曼光谱使用硅晶片在520.5 cm-1处校准。测试温度为室温,100-800 cm-1测试。
氢气程序升温还原(H2-TPR)测试过程如下,称取样品30 mg,在气体流量为30 mL/min N2条件下以10 ℃/min的升温速率升温至300 ℃,恒温保持40 min,然后降至室温,在30 mL/min H2/Ar (H2=5%)的气流下以10 ℃/min升温至850 ℃,检测器为TCD。
原位漫反射傅里叶变换红外(in-situDRIFTS)实验在装有Harrick Scientific漫反射附件的Nicolet iS 50红外光谱仪上进行。测试前,将样品置于配有ZnSe窗片的反应池中,N2(100 mL/min)氛围下在200 ℃吹扫60 min,待其降至室温。升温至70 ℃,第一,通入HCHO + N2吸附30 min;第二,N2吹扫15 min;第三,通入O2+ N2进行反应,采用OMNIC软件采集不同反应条件下的谱图。测试条件为通入0.1% HCHO、10% O2、N2作保护气,70 ℃时采集不同时间点的光谱。
1.3 催化剂的活性测试
催化剂活性测试在固定床石英管(i.d. = 6 mm)中进行。采用一定流量的氮气鼓泡37%甲醛溶液产生可控体积分数的HCHO/N2混合气体,0.25%HCHO、20%O2、N2为平衡气,混合气总流量为100 mL/min,催化剂用量200 mg。催化剂测试前先于10%H2/N2氛围下在150 ℃还原1 h。稳定性测试于80 ℃下进行30 h连续反应,气体组分为:0.2%HCHO + 20%O2+ N2,混合气总流量为100 mL/min。反应后的气体通入带TCD和FID检测器的9780型气相色谱在线分析,HCHO转化率用CO2的生成量评价,计算方法:
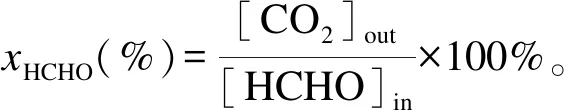
(1)
式中,[CO2]out:产物中CO2体积分数,[HCHO]in:进气中HCHO体积分数。
2 结果与讨论
2.1 活性测试
各催化剂的甲醛催化氧化活性测试结果见图1。
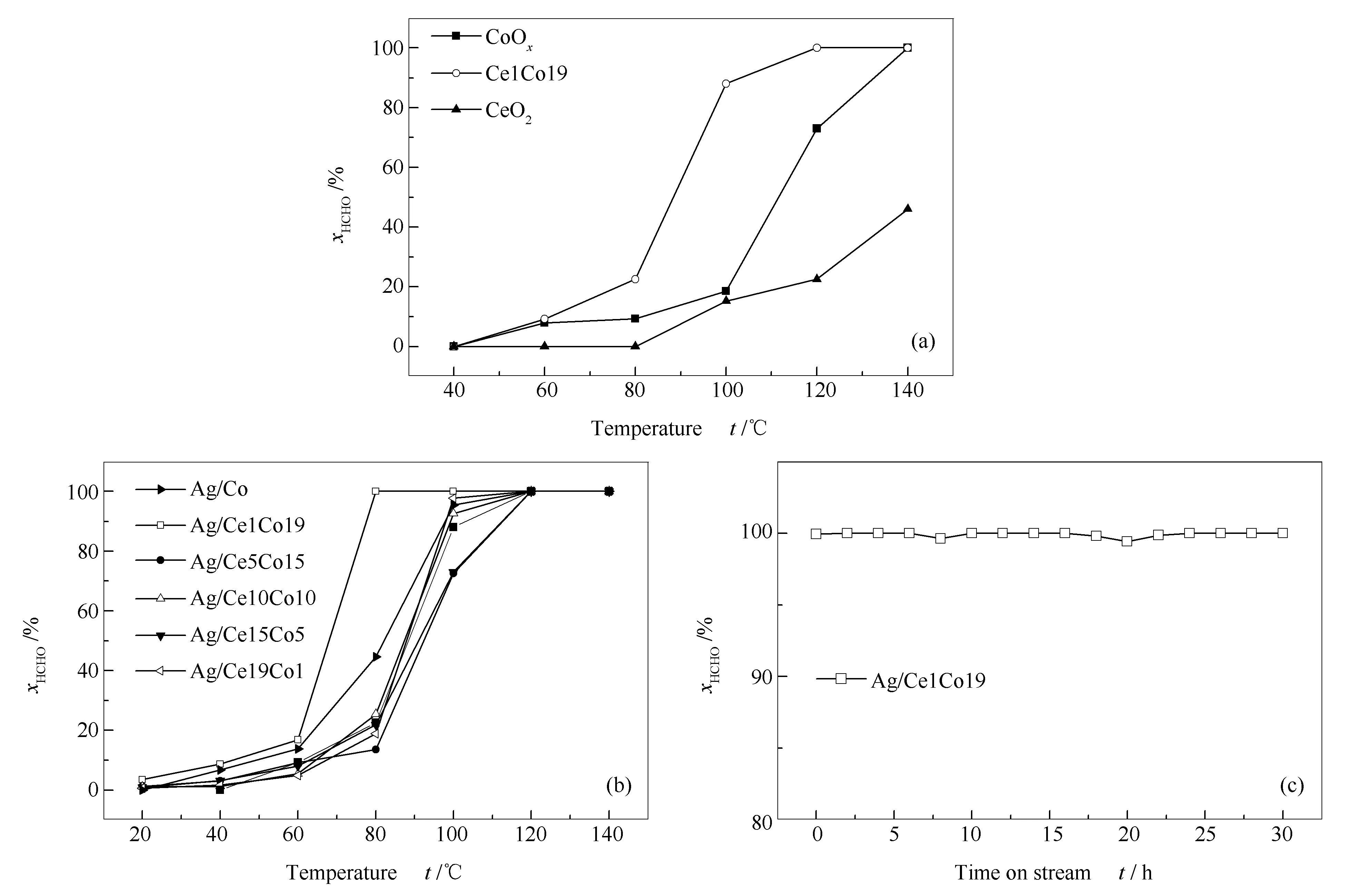
图 1 不同Ce/Co物质的量比对催化剂甲醛催化性能的影响
CeO2、CoOx、Ce1Co19氧化物单独作为催化剂的评价结果见图1(a),其中,纯CeO2显示出较差的催化活性,在140 ℃仅实现46%的甲醛去除率,而CoOx在相同测试条件下能够实现甲醛的完全转化。相比之下,Ce1Co19催化剂能够在120 ℃达到100%甲醛转化率。因此,不同Ce/Co比显著影响CeO2-Co3O4催化剂的甲醛催化降解效率。文献报道称Ag物种可以活化载体表面氧物种并促进其表面还原[15],同时,添加Ag物种导致反应气流中的氧分子有效活化更加显著[21]。如图1(b)所示,Ag加入后甲醛活性显著提升,证明Ag有效参与甲醛催化降解反应。各催化剂的t90大小依次为:Ag/Ce1Co19 (78 ℃) < Ag/Ce19Co1 (98 ℃) ≈ Ag/Co (98 ℃) < Ag/Ce10Co10 (99 ℃) < Ag/Ce5Co15 (112 ℃) ≈ Ag/Ce15Co5 (112 ℃)。其中,Ag/Ce5Co15和Ag/Ce15Co5催化剂显示出最差的活性,在120 ℃才能实现甲醛的完全转化。当Ce/Co物质的量比为1∶1时,显示出增加的HCHO催化活性,相比之下其t90提前了13 ℃。随着Ce或者Co含量的急剧增加,达到富Ce(Ag/Ce19Co1)或富Co(Ag/Ce1Co19)状态,催化剂活性进一步提高。Ag/Ce19Co1在100 ℃实现98%的甲醛转化,与纯Ag/Co显示出相当的活性。相比之下,Ag/Ce1Co19显示出最优异的低温催化氧化降解甲醛性能,在80 ℃即可实现甲醛完全转化。同时,Ag/Ce1Co19催化剂表现出优异的稳定性,能够在80 ℃下保持100%甲醛转化率达30 h,具体见图1(c)。这说明适量的CeO2加入有利于催化活性增加。由此可知,对于CeO2-Co3O4复合载体体系,适当的Ce/Co比对最终催化剂活性具有显著影响。总的来说,富Co状态下甲醛催化性能最优异,且其贵金属负载量(1% Ag)成本也低于大多数现有文献[13-15]。
2.2 XRD分析
各催化剂的XRD谱图见图2,用以揭示催化剂的结构特征。由图2可知,Ag/Co样品中位于19.0°、31.3°、36.9°、44.8°、55.7°、59.4°和65.2°处不同强度的衍射峰分别归属于Co3O4的(111)、(220)、(311)、(222)、(400)、(422)、(511)和(440)晶面(PDF#42-1467)。随着Ce的加入系列催化剂由CeO2立方萤石结构和Co3O4尖晶石结构混合组成,且Co3O4的结晶度相应变弱。随着Ce含量增加,位于28.6°、33.1°、47.5°、56.3°、69.4°、76.7°和79.1°处归属立方萤石结构CeO2的衍射峰峰强度逐渐增强,说明其结晶度不断增加。然而,当达到富Ce状态时,Ag/Ce19Co1催化剂对应的CeO2的衍射峰峰强度相对于其他增加趋势反而出现了下降,表明其结晶度降低,对应缺陷增多,更有利于催化活性。这可能是其在活性测试中活性优于其他较低Ce含量催化剂的原因。此外,所有样品中均未观察出明显的银物种衍射峰,可推测出低含量的银物种高度分散在催化剂表面而难以检测。
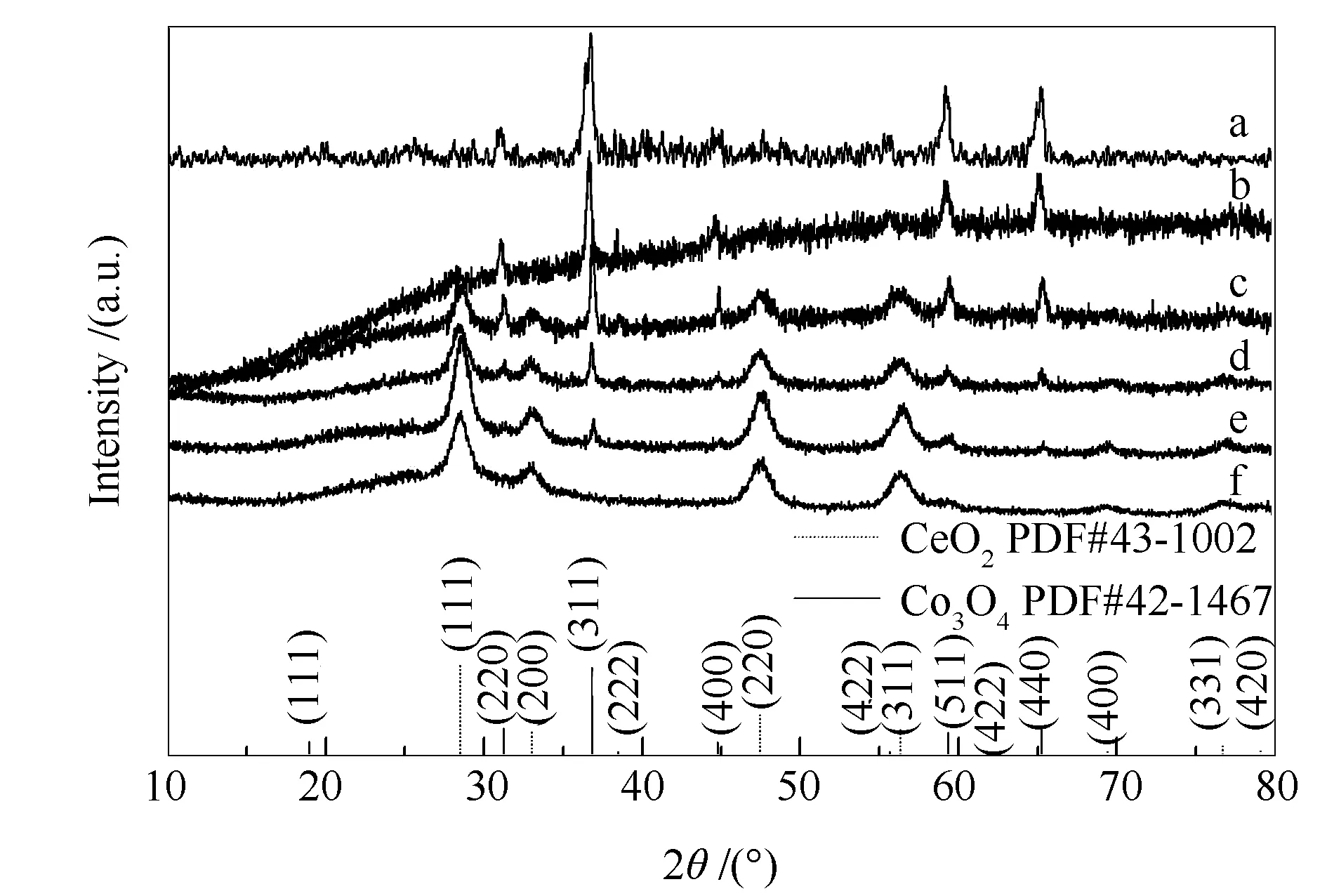
图 2 催化剂的XRD谱图
2.3 BET分析
各催化剂的N2吸附-脱附曲线见图3,观察到所有催化剂呈现出IV型吸附-脱附等温线,H4型滞回线,属于典型的介孔特征[22]且存在狭窄的裂隙孔。随着Co含量的增加,滞回线起点的相对压力p/p0逐渐增大,说明催化剂的介孔孔径逐渐增大。这与表1中的平均孔径结果相一致,其孔径大小随着Co含量的增加而增加,Ag/Co样品显示出最大的孔径(17.1 nm)。由此可知,不同Ce/Co比对催化剂的织构性质有着显著影响。表1汇总了不同Ce/Co比的Ag/CeO2-Co3O4催化剂的孔结构参数。由表1可知,系列催化剂的比表面积随着Co含量的增加而减小,反之,孔体积和平均孔径则随着Co含量的增加而增加。可能是由于Co的增加促进介孔尺寸增大而导致的。结合活性测试结果可知,织构性质并不是影响系列催化剂活性的主要因素。
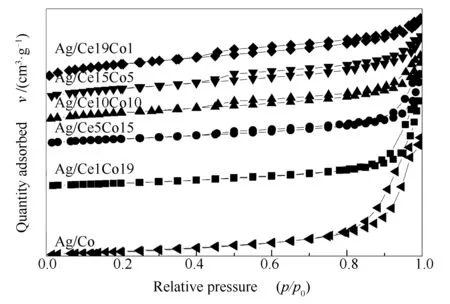
图 3 不同催化剂的N2吸附-脱附曲线
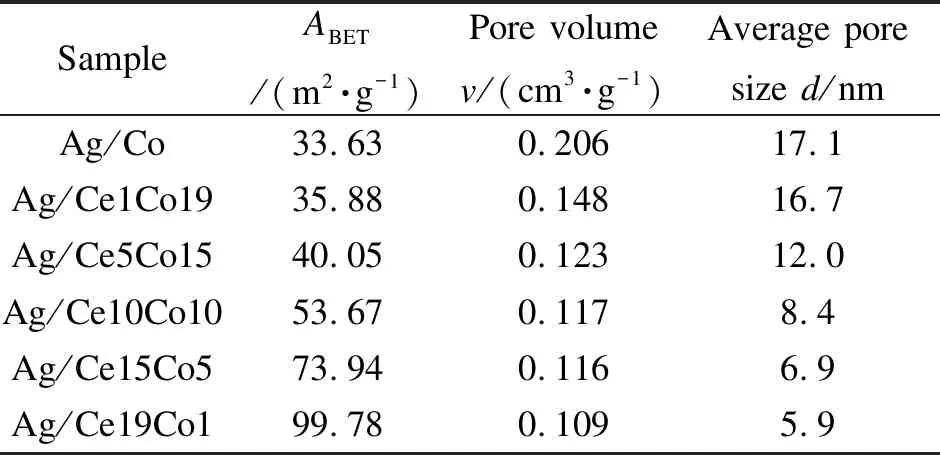
表 1 不同催化剂的孔结构参数
2.4 Raman分析
各催化剂结构通过Raman光谱技术进一步研究,测试结果见图4。由图4可知,所有催化剂的Raman谱图中均未发现Ag物种的衍射峰,表明系列催化剂中Ag物种无明显的拉曼散射信号,这与XRD结果一致。在Ag/Co催化剂上位于196、483、519、620和 689 cm-1波数附近分别对应于Co3O4中F2g(1)、Eg、F2g(2)、F2g(3)和A1g模式的伸缩振动[23]。研究发现,Co3O4的普通尖晶石结构中,Co2+和Co3+分别位于四面体和八面体位置[24]。其中,F2g和Eg模式与四面体和八面体位置的振动有关,A1g模式则对应于八面体位置的出现。随着Ce的加入观察到位于462 cm-1波数附近的峰属于CeO2中F2g模式的伸缩振动[25]。由图4可知,随着CeO2含量的增加,CeO2对应的散射峰明显增强,且随其强度的增加逐渐掩盖了位于475 cm-1附近归属于八面体位置振动的Co的散射峰。与此同时,相比Ag/Co催化剂,随着CeO2的加入,Co3O4对应的散射峰出现了红移,且随Ce含量的增加而减弱。其中,Ag/Ce1Co19催化剂的散射峰相对于其他催化剂出现了明显的红移,与其优异的活性结果一致。这可能是由于晶格缺陷导致的,它能够促进氧空位的形成从而促进氧气的移动[26,27]。通常情况下,表面上高氧迁移率和氧空位的存在可以改善氧化活性[28]。
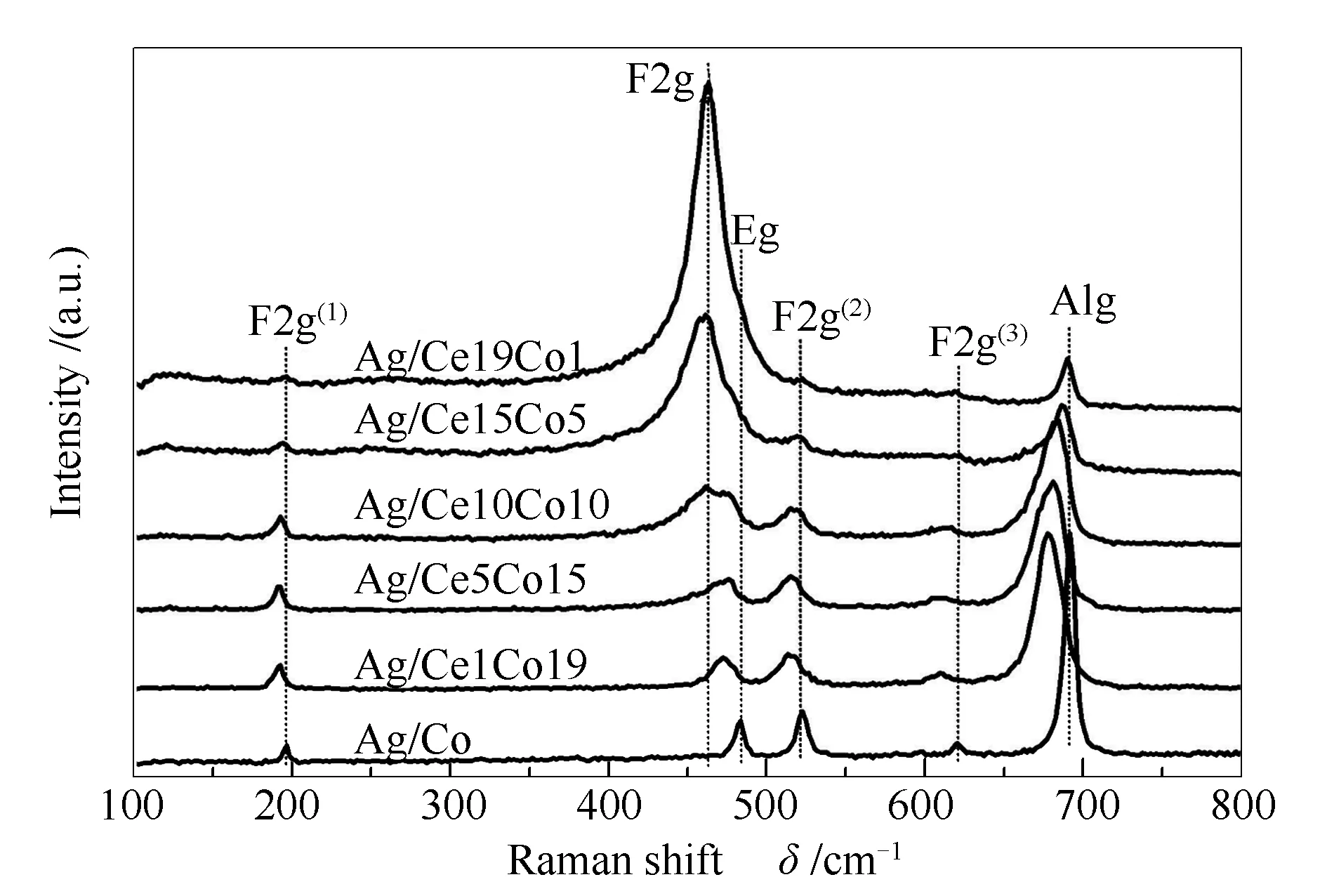
图 4 不同催化剂的拉曼光谱谱图
2.5 H2-TPR分析
为探究不同Ce/Co比催化剂的氧化还原性能,H2-TPR分析用来测定其氧化还原能力,所得结果见图5。系列催化剂在50-850 ℃呈现出还原峰。根据文献报道[17,29],Co3O4一般经过两步还原过程:第一步过程属于Co3+向Co2+的转化;第二步过程是Co2+还原为Co0。由图5可知,Ag/Co样品中位于251及350 ℃附近的两个还原峰归属于Co3O4的两步还原。其还原温度相比现有文献中的纯Co3O4对应还原温度都有明显提前[5,23,27],同时,随着CeO2的加入其还原温度逐渐向低温偏移,并随Ce含量的增加偏移愈加明显,说明Ce的存在有利于Co3+和Co2+的还原[28]。另外,出现位于100-150 ℃和高于700 ℃(插图)的新还原峰,分别归因于载体中CeO2表面的活性氧物种和CeO2中体相氧的还原[15,30]。Yu等[17]发现,Ag纳米粒子的存在能够促进CeO2的还原峰向低温偏移从而提高其氧化还原性能。与此同时,相应的H2消耗量也随着Ce含量的增加而增加,Ag/Ce19Co1催化剂表面存在大量的氧物种可能是其在活性测试中显示出仅次于Ag/Ce1Co19的原因。此外,结果表明,随着催化剂中Co含量逐渐增加,H2消耗量也随之增加,通常表面Co3+的含量与催化活性相关[31]。根据电中性原理,Co2+含量增加则表面氧空位含量增加,这与Raman结果一致。
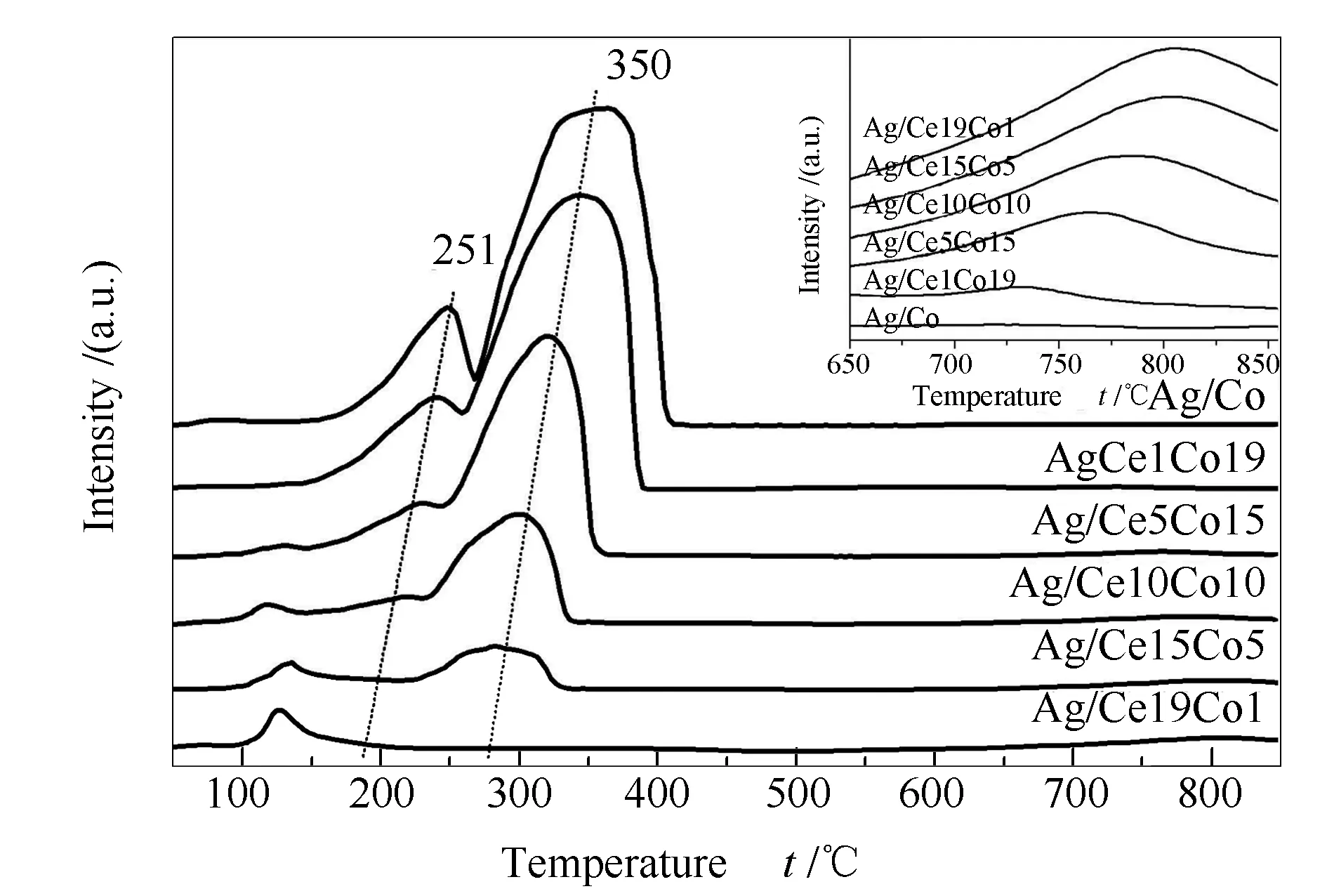
图 5 催化剂的H2-TPR谱图
2.6 原位漫反射傅里叶红外光谱研究
Ag/Ce5Co15和Ag/Ce1Co19在70 ℃下的原位漫反射傅里叶变化红外谱图(in-situDRIFTS)见图6。由图6可知,所有样品的in-situDRIFTS谱图均未发现与吸附甲醛相关的峰,说明吸附甲醛能够立即被氧化形成中间产物。在甲醛吸附阶段,随着时间的延长,各吸附峰逐渐增强,说明中间产物在催化剂表面的吸附量增大。由图6(a)可知,位于1062 cm-1处的吸附峰为甲二氧基(DOM)物种的C-O伸缩振动[31-33]。在1322和1363 cm-1处的吸收峰归属于甲酸盐物种(COO)对称伸缩振动[34,35];对应在1586 cm-1处的吸收峰则归属于甲酸盐(COO)的不对称伸缩振动[34],而位于2830和2948 cm-1处的吸收峰归属于甲酸盐C-H伸缩振动[36,37]。随着吸附时间的延长,所有吸附峰的强度都有所增强并随后保持稳定。此外,在3674 cm-1处出现了一个微弱的倒峰[31],这归因于表面羟基的消耗,以将吸附态的甲醛悉数转化为DOM和甲酸盐中间产物。与之相比,Ag/Ce1Co19催化剂表面的吸附中间产物也主要为DOM和甲酸盐物种。但是,Ag/Ce1Co19表面相关峰的吸附量明显降低,吸附时间达到1 min之后,才有DOM和甲酸盐中间体先后逐渐积累。另外,在1646和3470 cm-1处观察到新的吸收峰。根据文献,该吸附峰可归属于H2O的O-H伸缩振动[34,36]。说明在不通氧气的情况下,CeO2-Co3O4表面的氧物种直接参与甲醛的氧化同时生成CO2和H2O。
为了进一步探究反应中间体的演变过程,在吸附过程随后的通氧反应实验中,先通入N2吹扫15 min去除弱吸附的中间体,立即通入O2+ N2并采集不同反应时间的谱图。由图6(b)可知,DOM的吸收峰明显减弱,而甲酸盐物种则以微弱的趋势减少,这可能是由于Ag/Ce5Co15上的甲醛反应转化率在70 ℃低于20%导致的。说明甲酸盐物种的分解过程是甲醛氧化反应中的速控步骤。反观Ag/Ce1Co19催化剂,如图6(d)所示,随着氧气的通入,反应中间体的吸附量立即减少,并随反应时间的延长进一步减弱。可能是其表面的氧空位更有利于氧气分子的活化和移动,这与Raman分析结果相对应。经上述分析可知,Ag/Ce1Co19具有更好的吸附并转化甲醛的性能。结合活性测试结果证实了Ag/Ce1Co19催化剂表面存在更多的活性位点,使得吸附的甲醛物种与催化剂表面的活跃氧物种发生氧化反应,这也是Ag/Ce1Co19催化剂呈现最优活性的原因。
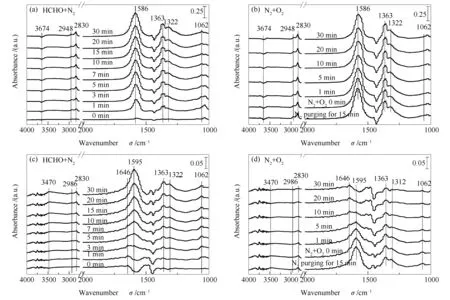
图 6 70 ℃下HCHO吸附(a)随后通氧反应(b)期间在Ag/Ce5Co15上及HCHO吸附(c)随后通氧反应(d)期间在Ag/Ce1Co19上获得的原位漫反射傅里叶红外光谱谱图
3 结 论
低Ag含量的Ag/CeO2-Co3O4催化剂表现出较好的低温催化氧化降解甲醛性能,并且不同Ce/Co比对催化剂的活性影响显著,其中,Ag/Ce1Co19在80 ℃实现了甲醛的完全转化。结果表明,富Co状态下,少量Ce的加入有效地提高了晶格缺陷,促进氧空位的形成,从而优化甲醛低温催化氧化降解性能。此外,in-situDRIFTS研究结果表明,氧空位有利于促进反应过程中氧气分子的活化和移动,载体表面氧物种能够直接氧化甲醛形成DOM和甲酸盐物种反应中间体,且甲酸盐物种的分解是甲醛催化氧化过程中的速控步骤。
猜你喜欢
真空与低温(2022年6期)2023-01-06 07:33:20
生物学通报(2021年4期)2021-03-16 05:41:26
现代塑料加工应用(2021年5期)2021-02-28 08:18:04
江苏安全生产(2020年1期)2020-03-16 12:57:50
劳动保护(2018年8期)2018-09-12 01:16:18
中国蜂业(2018年4期)2018-05-09 06:25:08
当代化工研究(2016年6期)2016-03-20 16:21:46
发明与创新(2015年30期)2015-02-27 10:39:49
中央民族大学学报(自然科学版)(2014年3期)2014-06-09 08:54:31
中国质量与标准导报(2014年6期)2014-02-28 22:24:11
