
二元硼铝复合团簇B7Al20-和B7Al30-的结构、成键和芳香性研究
2019-05-31 01:13王渊冯林雁王康翟华金
山西大学学报(自然科学版) 2019年2期
王渊,冯林雁,王康,翟华金
(山西大学 分子科学研究所,山西 太原 030006)
0 引言
硼是元素周期表中的一种独特元素,具有丰富多样的化学性质。Lipscomb在硼烷研究中提出的三中心两电子 (3c-2e) 键在现代化学键理论中扮演着重要角色。在过去30年间硼纳米团簇受到了科学界的广泛关注[1-13],特别是一系列旨在探索硼团簇结构和成键的实验和理论研究开辟了平面硼 (planar boron) 的新大陆。研究发现多达40个原子构成的硼阴离子团簇均具有平面或准平面结构,而硼阳离子团簇在16个原子附近观察到平面向管状结构的转变。硼团簇的平面性源于其缺电子性。平面硼团簇除去外围B—B单键之外,还存在离域π/σ键[9],导致该体系具有芳香性、反芳香性和冲突芳香性。此外,硼化学的新发展还包括动力学流变性纳米转子[14-19]、硼羰基化合物[20-21]、硼球烯 (borospherenes)[13,22-23]和硼烯 (borophenes)[12,24-25]等等。
通过掺杂第二种元素可调整硼团簇的结构、电子特性和化学键,由此形成硼基二元复合纳米团簇。基于这些二元团簇可调控体系的电子数目,进而改变团簇的成键性质和结构。迄今为止,金属掺杂硼团簇已得到广泛研究。Wang与合作者结合光电子能谱和量子化学计算报道半三明治PrB7-、CoB12-和RhB12-团簇[26-27]。该小组还研究过渡金属掺杂B8、B9和B10团簇,发现新颖的高配位平面分子轮,最高配位数为10[28-29]。Dong等[30]预测管状Li2B12和笼状Li3B12团簇。2016年Jian等[31]报道以Mn为中心的双环管状MnB16-团簇。Popov等[32]在实验上表征了高配位的鼓状CoB16-团簇。对于更大尺寸的掺杂团簇,CoB18-、FeB18和FeB20均为类富勒烯结构[33]。最近Zhai与合作者设计了金属掺杂的硼基三维纳米转子,发现有趣的动力学结构流变性现象。例如Mg2B8团簇的动力学行为酷似指南针[17],而Be6B11-和Be6B102-团簇[18-19]则展示双重动力学模式,即所谓“亚纳米类地月体系”。
倘若金属掺杂剂是铝,会使硼团簇结构发生什么样的变化?会如何影响硼团簇的化学键和芳香性?这些问题在团簇科学中值得深入探索。首先,虽然硼原子和铝原子是等价电子的,但它们的团簇结构却不相同,且平面结构为纯硼团簇独有[9]。其次,铝团簇是全金属芳香性体系的典型代表[34-35],将铝硼混合在一个二元体系电子离域性如何变化是一个有趣而待探索的问题。第三,考虑到硼是缺电子元素而铝是金属 (电子供体),铝掺杂应当有助于弥补硼的缺电子性,具体探索这一过程是有意义的。迄今为止,二元硼铝团簇已有一些文献报道[36-38],但是对于多个铝原子掺杂的二元硼铝团簇人们仍然知之甚少。
本工作我们将研究二元硼铝团簇B7Al20/-和B7Al30/-系列。我们的理论工作包括使用Coalescence Kick(CK)程序进行全局极小结构(global minimum,GM)搜索[39-40];基于B3LYP方法的几何结构优化和CCSD(T)单点计算;通过正则分子轨道(canonical molecular orbital,CMO)、适应性自然密度划分(adaptive natural density partitioning,AdNDP)[41]和自然键轨道(natural bond orbital,NBO)程序[42]等对四个GM体系进行成键分析。理论结果表明B7Al20/-和B7Al30/-团簇的GM结构不同于等电子体的B90/-和B100/-团簇。B7Al20/-和B7Al30/-团簇最优结构分别含有B7七元环或六配位轮状B7基盘,这些纳米结构是硼团簇和低维硼纳米材料的重要构筑单元。铝原子掺杂剂在二元硼铝团簇中发挥灵活的作用,有助于调整体系的电子计数,使之符合 (4n+2) Hückel芳香性规则。成键分析结果显示,四个硼铝团簇都具有双重π/σ芳香性 (6π和6σ电子计数)。这一成键特征可能具有普遍意义,将有助于指导新型芳香性硼铝团簇的理性设计。
1 理论方法
本工作采用全局CK搜索及手工搭建确定B7Al20/-和B7Al30/-团簇的GM结构。CK计算使用B3LYP方法[43-44]和3-21G基组。其后在B3LYP/6-311+G(2df)水平下对低能量异构体进行结构优化和频率计算,并将所得优化结构在CCSD(T)/6-311+G(2df)[45]水平下进行单点能量校准。最优结构确认对应于势能面上的能量极小点。使用CMO、AdNDP和NBO程序阐释体系的成键特性。所有计算均使用Gaussian 09程序包完成[46]。使用Molekel程序[47]对分子结构和化学键实现可视化。
2 结果
2.1 B7Al2和B7Al2-
计算结果表明,B7Al2(C1,2A)和B7Al2-(1,D7h,1A1′)团簇的GM结构一致。鉴于它们的相似性,我们将主要描述闭壳层结构B7Al2-(1) (图1(a))。结构1具有D7h对称性,由七边形B7环和中心Al2单元组成。其中两个铝原子分别位于B7环中心轴上方和下方,每个铝原子距离B7环平面约1.40 Å。根据Pyykkö建议的共价原子半径[48],B—B和B=B键长的上限分别为1.70和1.56 Å。因此1中B—B键长1.57 Å明显短于单键(接近双键),表明除去典型B—B两中心两电子(2c-2e)Lewis键外,团簇1中的B7环还具有离域键特征。Al—Al间距2.79 Å远超过单键键长上限2.52 Å,暗示它们之间可能没有定域键,共价作用很弱。B—Al间距为2.28 Å(未在图中表示),长于单键(2.11 Å)。中性B7Al2团簇的GM结构为C1(2A),但其实际上为D7h对称性,B—B、B—Al和Al—Al间距分别为1.58、2.23和2.57 Å(图1(a))。
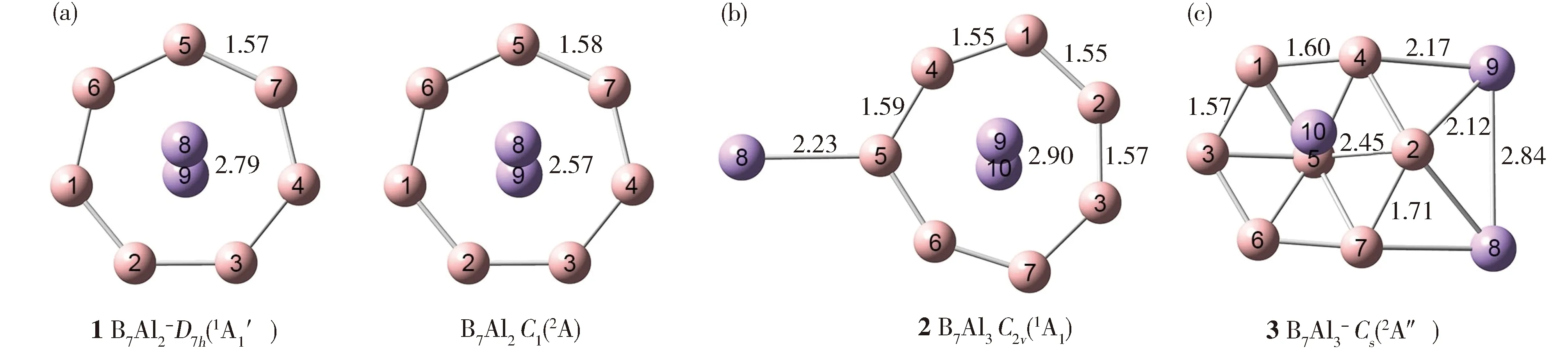
(a) B7Al2-(1,D7h,1A1′), B7Al2(C1,2A) and (b) B7Al3(2,C2v,1A1) clusters,as well as hexagonal B7disk based (c) B7Al3-(3,Cs,2A″) cluster. Selected bond distances are labelled in Å.The B atoms are shown in pink and Al in purple.Fig.1 Optimized global-minimum (GM) structures at the B3LYP/6-311+G(2df) level for heptagonal B7ring based(a) B7Al2-(1)和B7Al2;(b) B7Al3(2); (c) B7Al3-(3)。硼原子为粉色,铝原子为紫色,键长单位为Å图1 在B3LYP/6-311+G(2df)水平下优化的全局极小结构
2.2 B7Al3和B7Al3-
B7Al3团簇的GM结构为2(C2v,1A1) (图1(b))。团簇2也含有七边形B7环,从几何上可以团簇1为基础构建,即将第三个Al原子以端基形式连接到硼环上。结构2的B—B键长范围为1.55~1.59 Å,具有双键特征。与团簇1相比中心Al—Al间距(2.90 Å)长于团簇1,表明2的Al—Al成键作用更弱。末端B—Al键长(2.23 Å)长于单键(2.11 Å),主要是离子键,辅以微弱共价键(见下文)。Al9/Al10与B7环之间的B—Al间距为2.27~2.36 Å(未标出),与B7Al2-(1)类似。
B7Al3-团簇的GM结构为3 (Cs,2A″) (图1(c)),3在结构上明显不同于1和2。团簇3具有六配位B7盘状结构,三个Al原子分别位于硼环外侧和环上方。中心B原子高于环平面0.45 Å。团簇3中B—B键可分为两类,即较短的1.57/1.60 Å与较长的1.71 Å。前者接近于双键,后者可视为单键,这表明桥连的Al原子削弱了与之相邻的B—B键。由于径向B—B连接仅存在离域键,因此其键长略长于单键(1.68-1.72 Å,未标出)。拉长的径向B—B间距也导致团簇结构的非平面性。虽然桥连B—Al键(2.12/2.17 Å)接近单键,但这并不意味着其间存在传统Lewis键,见后面讨论。相比之下,中心B5—Al10间距更长(2.45 Å)。
2.3 韦伯键级和自然原子电荷
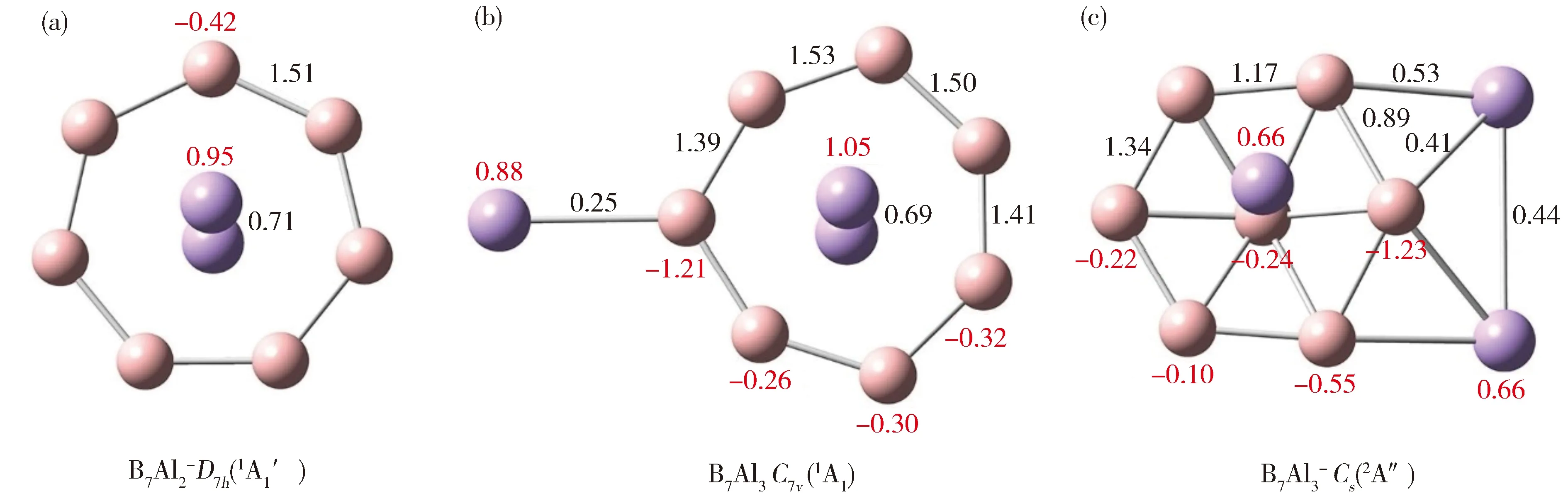
(a) B7Al2-(1);(b) B7Al3(2);(c) B7Al3-(3)Fig.2 Calculated Wiberg bond indices (WBIs; in black color) and natural atomic charges (in |e|; red color) from the natural bond orbital (NBO) analyses.图2 团簇1,2,3的韦伯键级 (黑色) 和自然电荷 (红色)
基于NBO分析我们计算了GM结构1-3的韦伯键级(Wiberg bond index,WBI)和自然原子电荷。计算结果总结如下:(1)外围B环(七边形或六角形)B—B键的WBI大于1。这与平面硼团簇中外围B原子同时参与2c-2e定域键和π/σ离域键的基本认知相一致[9]。而在团簇3中连接桥Al的两个B—B键级为0.89,这里B—B成键不再以简单的2c-2e σ单键存在;(2)由于存在离域π/σ键,3中径向B—B键的WBI为0.56-0.64(图中未标出);(3)在团簇1和2中,中心Al—Al键的WBI分别为0.71和0.69,这似乎与它们的原子间距 (2.79和2.90 Å) 相矛盾。事实上它们远大于Al—Al单键 (2.52 Å),暗示Al—Al的韦伯键级值可能是一个假象;(4)2中末端B—Al键级较低(0.25),与其键长似有矛盾,主要原因是B—Al之间存在较强的离子键作用。同样,3中悬浮在B7上方的Al与中心B形成的键级也较小(0.21);(5)桥连的Al—Al和B—Al键级分别为0.44和0.53/0.41,这表明Al—Al和B—Al有一定的共价作用。
关于自然原子电荷,团簇1-3中硼原子都带有负电荷(-0.10 ~-1.23 |e|)。由于极化作用,与端铝或与桥铝相连的硼原子带有大量的负电荷。另一方面,每个铝原子都带有正电荷(+0.66 ~+1.05 |e|)。我们发现保留了3s2孤对电子的铝原子似乎带有较少的正电荷。
3 分析与讨论
3.1 硼铝团簇B7Aln0/-(n=2, 3)成键分析
B7Al2和B7Al2-(1)团簇的GM结构类似,而B7Al2-(1)为闭壳层体系,因此我们主要讨论团簇1的成键。如图3所示,团簇1有28个价电子,占据14个价轨道。根据其原子轨道成分可将分子轨道分为四个子集。子集(b)有7个CMO,它们严格遵循轨道构建原则,从下到上依次含有0,1,2,3个节面,主要由硼的2s原子轨道组成。这些CMO线性组合可还原B7环的7个B—B Lewisσ单键。子集(c)中3个价轨道类似于苯的离域π轨道,符合(4n+2) Hückel规则。因此,团簇1具有π芳香性。同样地,子集(d)在空间分布上类似子集(c),但(d)的本质是σ轨道,由硼原子径向2p原子轨道组成,这些σ轨道也符合(4n+2) Hückel规则,赋予该团簇σ芳香性。子集(a)由Al 3s/3p原子轨道组成,两个铝原子间形式上形成σ键。基于以上成键分析,两个Al原子共有4个价电子以共价键或离子键的形式参与二元团簇体系离域键。NBO结果表明 (图2(a)),1中B—Al键是一半共价键一半离子键(轨道成分分析表明团簇1和2中Al2单元分别贡献给π键1.35和1.25 |e|,由此可见B—Al键具有混合的离子键和共价键特征)。团簇B7Al2与B7Al2-(1)相同,但前者是开壳层结构,比后者少一个价电子。
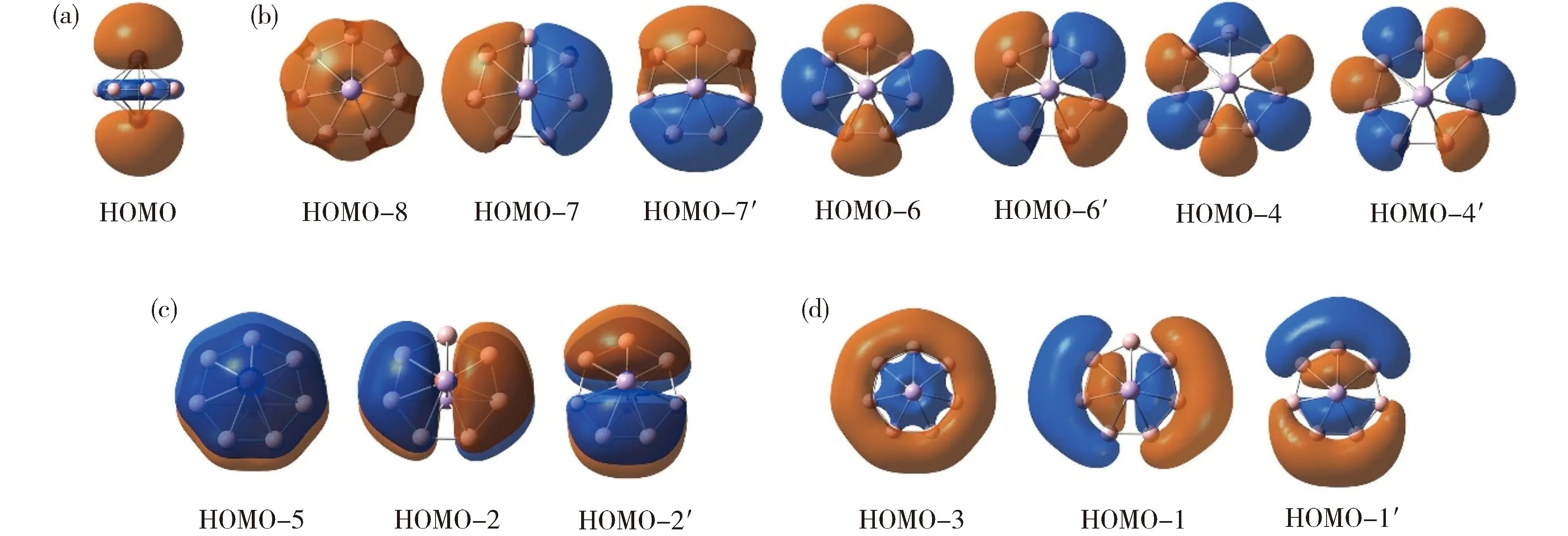
(a) One two-center two-electron (2c-2e) CMO for a formal Al—Alσbond;(b) Seven CMOs for peripheral 2c-2e B—Bσbonds in the B7ring;(c) Three delocalized π CMOs (π sextet);(d) Three delocalizedσCMOs (σsextet).Fig.3 Canonical molecular orbitals (CMOs) of B7Al2-(1) cluster.The CMOs are sorted to four subsets on the bases of their constituent atomic orbitals (AOs)(a)Al—Al “形式”σ键;(b)7个B—B 2c-2eσ单键;(c)3个离域π轨道;(d)3个离域σ轨道图3 团簇1的分子轨道分析
AdNDP分析可以直观地再现上述CMO分析结论。图4为团簇1的AdNDP方案:(a)展示形式上的Al—Alσ键,(b)为7个B—B单键,(c)和(d)分别为3个离域π键和3个离域σ键。这一成键图像确认团簇1的双重π/σ芳香性。

Fig.4 Bonding pattern of B7Al2-(1) based on the adaptive natural density partitioning (AdNDP) analysis. Occupation numbers (ONs) are shown.图4 团簇1的AdNDP成键图像。轨道占据数(ON)已列出
在建立B7Al2-(1)的成键图像之后,团簇B7Al3(2,C2v,1A1)的化学键就容易理解得多。从结构上看,B7Al3(2)以1为基础构建,第三个铝原子加在硼环外围形成端基。NBO数据表明团簇2可近似看作Al+[B7Al2]-(图2(b))。因此,B7Al3(2)的成键方式与B7Al2-(1)相似,且前者是基于后者的电荷转移化合物。团簇2的CMO证实上述想法(图5),除末端铝原子3s2孤对外 (HOMO—8;见图5(a)),其余CMO与团簇1一一对应(图3)。必须指出,我们没有找到与端基B—Al键对应的CMO,即该端基由离子键作用所主导。团簇2的AdNDP方案完美地重复上述CMO分析结果(图6)。
(a) One Al 3s2lone-pair;(b) One two-center two-electron (2c-2e) CMO for formal Al—Alσbonding;(c) Seven CMOs for peripheral 2c-2e B—Bσbonds in the peripheral B7ring;(d) Three delocalized π CMOs;(e) Three delocalizedσCMOs.Fig.5 Pictures of canonical molecular orbitals (CMOs) of B7Al3(2) GM cluster(a)1个Al 3s2孤对,(b)形式上的Al—Alσ键,(c)7个B—Bσ单键,(d)3个离域π轨道,(e)3个离域σ轨道图5 团簇2的分子轨道分析
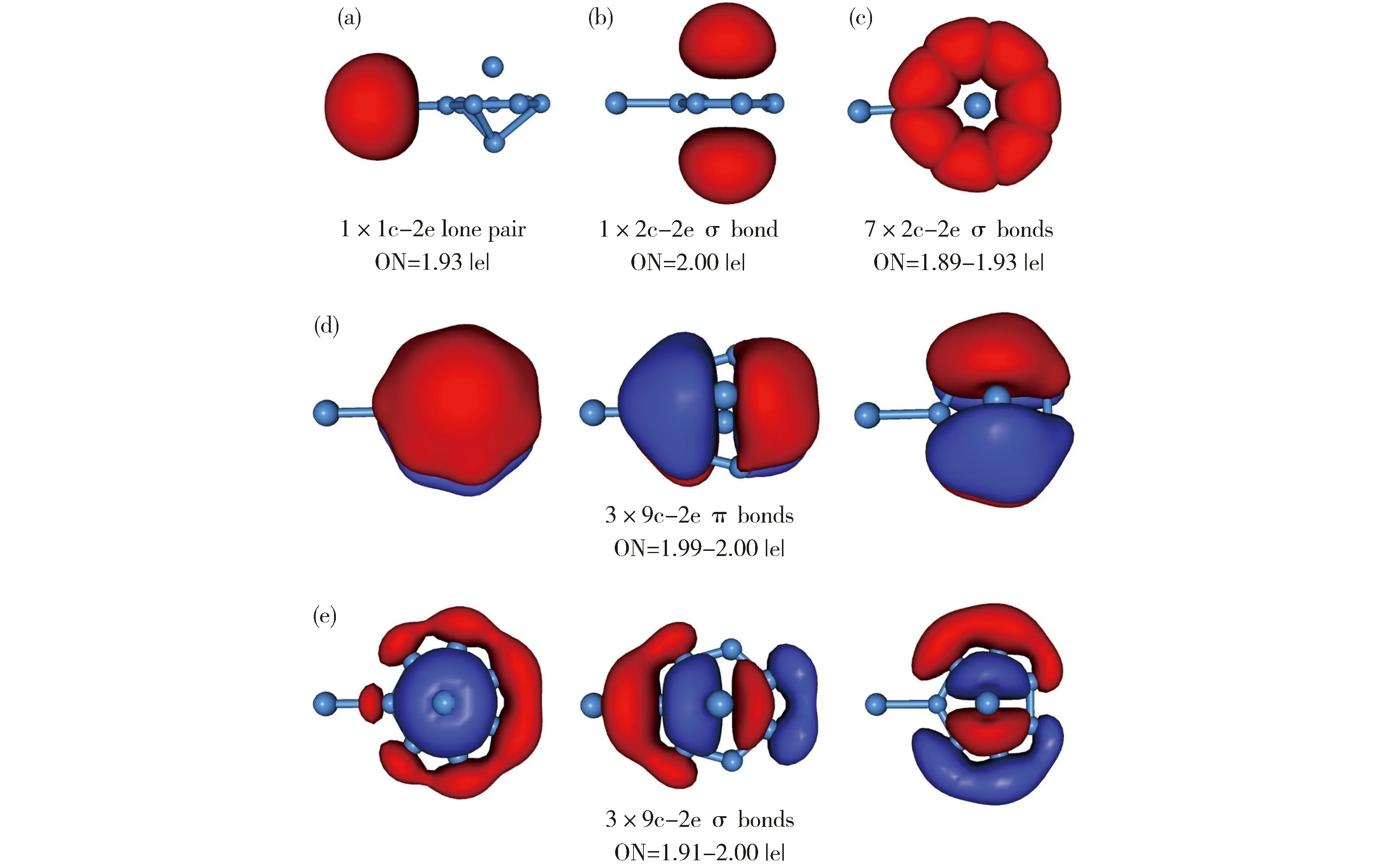
Fig.6 AdNDP bonding pattern of B7Al3(2). ONs are shown.图6 团簇2的AdNDP成键图像,轨道占据数(ON)已列出
有趣的是,当添加第三个Al原子、形成B7Al3-(3)团簇时,其GM结构发生显著变化 (图1(c))。团簇3是31个价电子的开壳层体系,其16个价层CMO如图7所示,其中HOMO是单占据轨道。图7(a)中3个CMO的主要成分是铝原子的3s/3p,共消耗5个价电子。这一结果表明体系中实际上仅有26个电子用于成键。其中子集(b)中7个CMO对应于外围原子的成键。如子集(c)(d)所示,除电子云略有畸变外,团簇3的离域轨道也与1和2相似。注意到它们的GM结构不同(图1)。作为技术说明,根据轨道构建准则,团簇3中的子集(b)可定域为2c-2e/3c-2e键。虽然团簇3是六配位硼环,但由于两个桥铝原子也参与外围成键,因此存在七个定域(或者准定域)σ键。其直观的AdNDP图像如图8(b)所示。
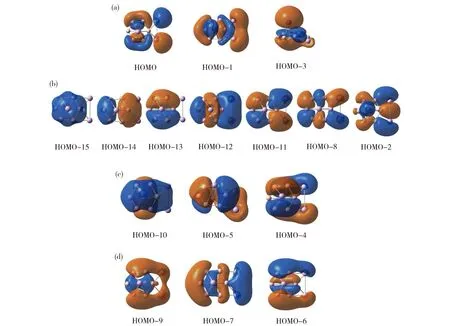
(a) Three CMOs primarily for Al 3s2lone-pairs (or nonbonding); HOMO is half occupied;(b) Seven CMOs for the peripheral (localized or island) σ bonds;(c) Three delocalized π CMOs;(d) Three delocalized σ CMOs.Fig.7 Pictures of CMOs of B7Al3-(3) GM cluster(a)3个Al 3s2孤对或非键轨道 (HOMO为单占据),(b)7个外围σ键(定域键),(c)3个离域π轨道,(d)3个离域σ轨道图7 团簇3的分子轨道分析
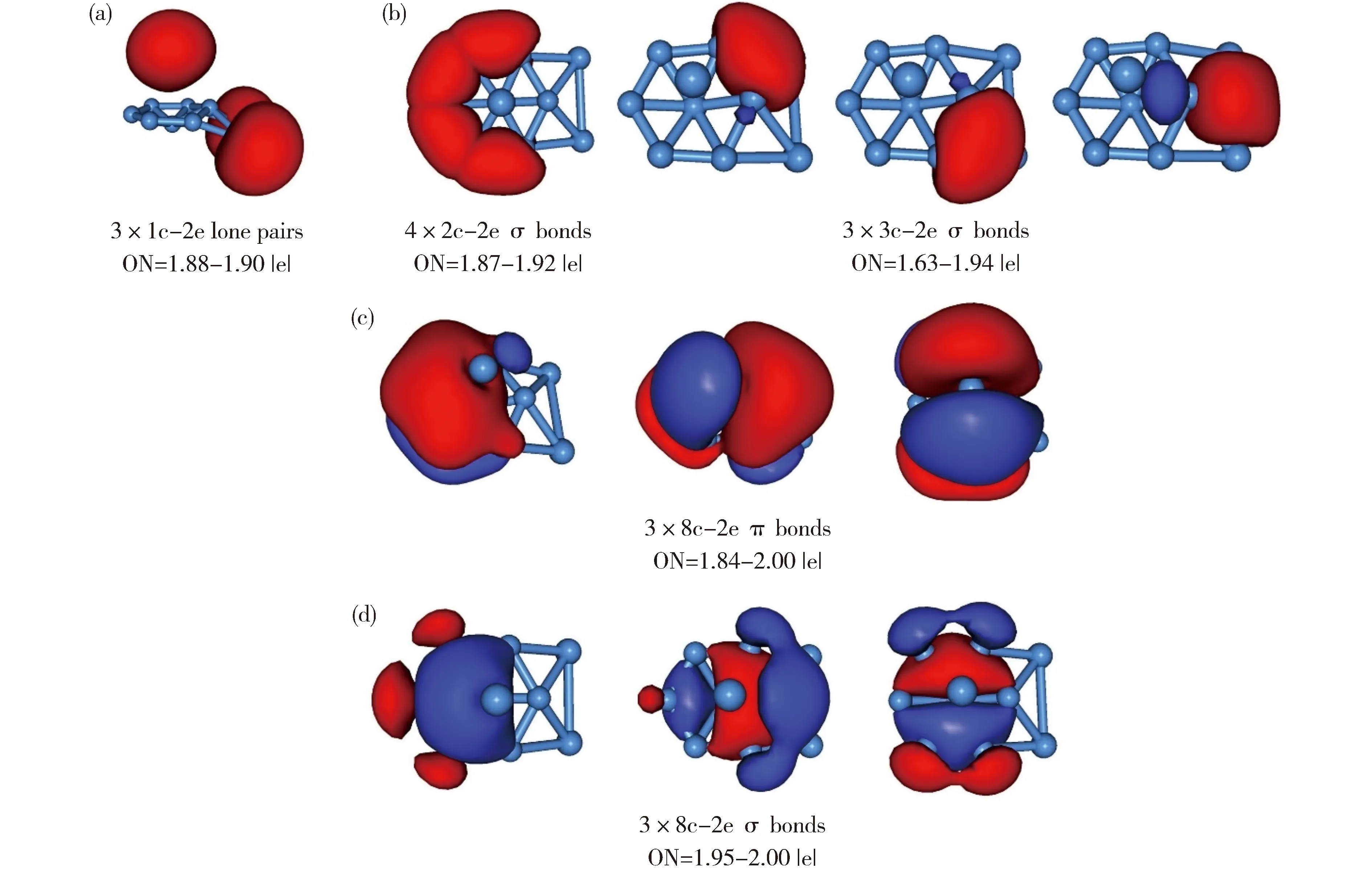
Fig.8 AdNDP bonding pattern of B7Al3-(3).Since the species is open-shell, one extra electron is added in the analysis. ONs are shown.团簇3是开壳层结构,故AdNDP分析时添加一个额外电子图8 团簇3的AdNDP成键图像,轨道占据数(ON)已列出。
综上所述,团簇1-3都具有双重芳香性。我们认为这种独特成键模式是团簇1-3稳定性的基础。团簇1-3中双帽铝原子和桥铝原子以共价键的形式参与离域π/σ成键,而团簇2中端基铝原子和团簇3中面上铝原子则以离子键为主。
文献中已报道BnAl-(n=7-9)[36-38]等二元硼铝团簇和纯硼团簇Bn-(n=8-10)[6-7]。B7Al-和B8Al-团簇[38]都是具有一个中心硼的盘状结构,铝原子位于中心硼的上方。B8-和B9-团簇[6]则分别为七配位和八配位平面分子轮状结构。B9Al-和B10-团簇[7,37]均为二维片状结构。显然,上述团簇结构与图1所示B7Aln0/-(n=2, 3)团簇GM结构迥异,无须赘述。
3.2 B7Al30/-团簇中七元硼环和六配位盘状结构的竞争
B7Al3(2) 和B7Al3-(3)仅相差一个电子,但它们的结构却完全不同(图1)。团簇2具有七边形B7环,并以团簇1为基础通过硼环末端连接第三个铝原子构建。实际上,如3.1节所述B7Al3(2)是电荷转移化合物Al+[B7Al2]-。而团簇B7Al3-(3)含六配位B7盘状结构,它由半夹心B7Al-结构、通过平面内桥接两个铝原子而形成。由于3中铝原子倾向于保留一定数量的3s电子(包括3s2孤对),三个铝原子共提供四个电子用于二元体系的共价键,因此六角形B6环转变为扭曲的具有七个定域键的B6Al2系统。团簇3中的B7片段与独立的B73-体系并不相同[26]。团簇3的B7片段应视为B75-(来自于三个铝原子的4个电子加上一个额外负电荷),所以不应是B73-。只有当3形式上处于B75-电荷态时我们才能解释7个2c-2e/3c-2eσ键以及离域6π/6σ电子计数。但是对于裸B75-团簇,形成7个外围σ键可能会存在困难。因此,我们认为B7Al3-(3)和B82-团簇是比较理想的等价体系。

Relative energies are shown in eV at the B3LYP/6-311+G(2df) level.Left side: heptagonal B7ring based isomers (2 and 5).Right side: hexacoordinate B7disk based isomers (4 and 3).Fig.9 A schematic diagram for energetics relationship of two-types of isomeric structures of B7Al30/-clusters in two charge-states.相对能量为B3LYP/6-311+G(2df)水平下计算结果(单位:eV)图9 两种B7Al30/-团簇构型的能量关系图
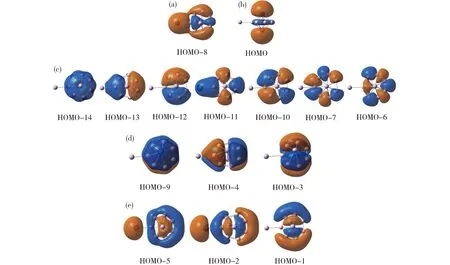
为什么一个电子使团簇结构发生从B7Al3(2)到B7Al3-(3)的变化?为了解决这个问题,我们分析两种中性和阴离子态的七元环(2/5)和六配位盘状(4/3)结构在B3LYP水平的能量关系,如图9所示。中性B7Al3团簇倾向于七元环C2v结构2,六配位型异构体4 (Cs,3A)比最优结构2高出0.39 eV (图9)。当分别添加一个电子到2和4时,两种结构分别被稳定至5和3。稳定程度取决于电子亲和能的大小。异构体4的电子亲和能为2.80 eV,远高于团簇2的1.81 eV。电子亲和能的差值 (0.99 eV) 显著提高团簇3的相对稳定性,这足以翻转中性团簇2相对于4的能量优势。因此,具有六配位盘状结构的团簇3成为阴离子GM,比七元环异构体5稳定0.60 eV。此外,由于额外的电子填到HOMO(最高占据轨道),因此该结果与团簇3和5的HOMO轨道性质相关(图10)。对于B7Al3-(3),HOMO轨道中两个桥铝原子共贡献55% (包括31% Al 3s和24% Al 3p),其余成分为B 2p轨道。相比之下,异构体5的中心Al2单元共贡献82% (包括36% Al 3s和46% Al 3p),两个铝原子间成反键形式。我们认为在团簇3中硼组分参与HOMO轨道有利于稳定该阴离子结构,这也是结构4的电子亲和能较大的原因。
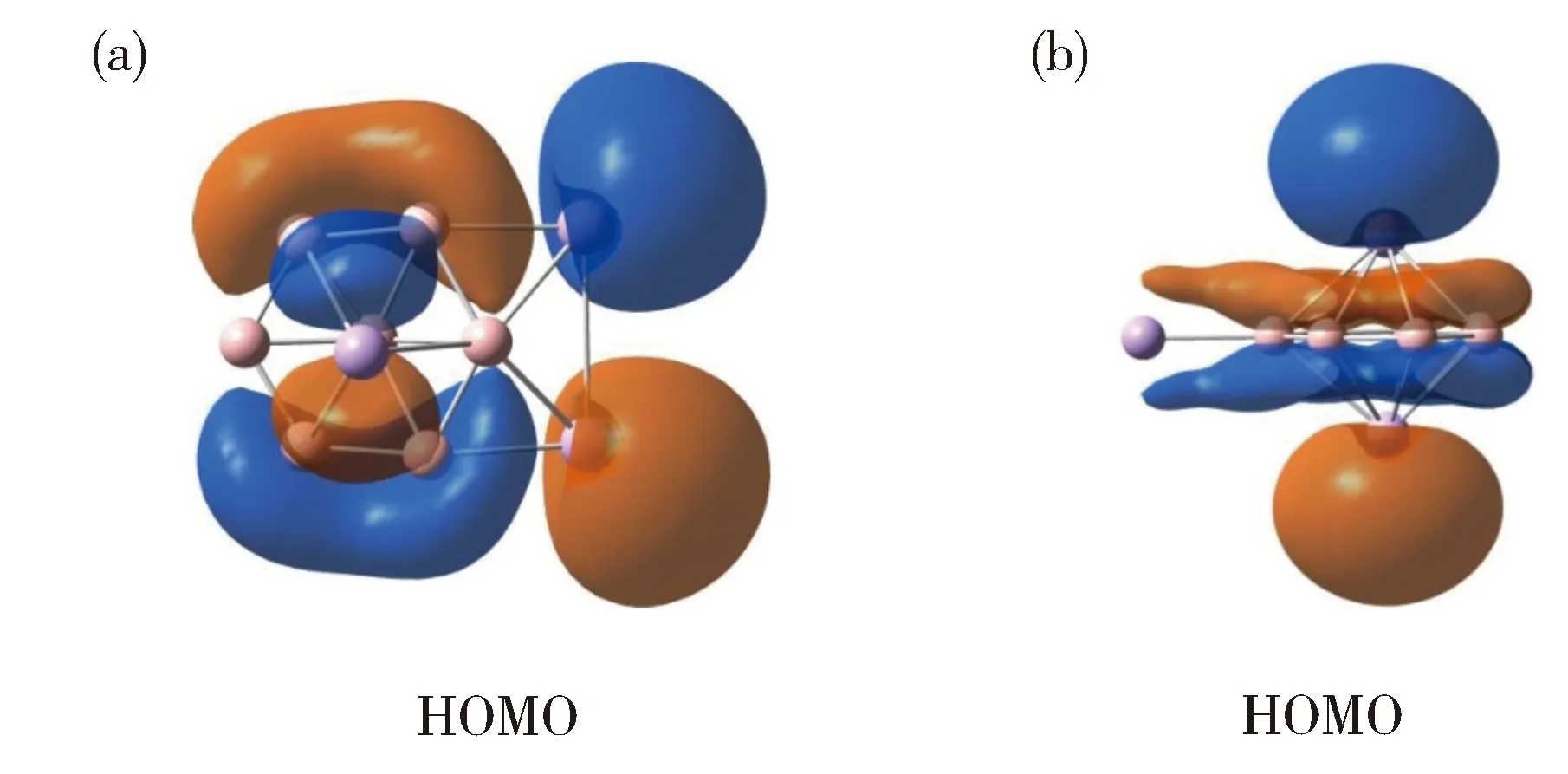
Fig.10 Comparison of the HOMO of(a) B7Al3-(3) and (b) B7Al3-(5) clusters.图10 团簇3 (a)和5 (b)的HOMO轨道比较
3.3 BmAln0/-团簇中的等价Al/B取代
等电子Al/B取代激发人们对于二元B—Al团簇[36-38]的研究兴趣。但是,我们这项工作表明“等价取代”的想法并不完全正确。在B7Al20/-和B7Al30/-团簇(1-3)中铝原子可分为四类:(1)七边形硼环上以双帽形式存在的Al2单元;(2)端基B—Al链接;(3)六配位B7面上铝原子;(4)桥基铝原子。其中类别(1-3)在纯硼团簇中不存在[1-13]。虽然类别(4)是硼团簇的重要组成单元,但B7Al3-(3)与B9Al-或B10-团簇[7,37]在结构上明显不同。上述事实表明,等电子Al/B取代并不是真正意义上的等价取代,它不能解释B7Al20/-和B7Al30/-团簇与其相对应B9-和B10-团簇[6-7]GM结构的差异。
根据成键分析,我们对B7Al20/-和B7Al30/-团簇中的Al/B取代有深入的认识。B—Al键兼具离子键/共价键的性质,其共价程度与铝原子的类型相关。对于前述类别(2)和(3)主要表现为一个电子的电荷转移,几乎没有B—Al共价键。对于类别(1)和(4),铝原子与硼原子共同参与共价π/σ键。需要强调的是,即使是后一种情况等价Al/B取代的概念对B7Al20/-和B7Al30/-团簇仍然无效。
一个关键原因是团簇1-3中铝原子保留了一定数量的3s/3p电子。例如,1中双帽Al2单元保留两个电子(图3(a))。2中末端铝原子保留3s2孤对,同时双帽Al2单元仍然保留两个电子(图5)。3中三个铝原子仅提供四个电子与硼原子键合(图7)。但对于纯硼团簇来说,所有价电子都参与成键。因此,B7Al20/-和B7Al30/-团簇与B9-和B10-团簇[6-7]并非等价体系。具体地说,B7Al30/-(2和3)分别与B100/-相差4和5个电子,2和3都是26电子体系(只计算参与成键的电子数)。从这一点来看,B7Al3-(3)团簇实际上是B82-的等价物种(参见3.2节讨论)[6],它们均为26电子体系,具有7个外围Lewisσ键和6π/6σ离域电子。这个类比有点令人惊讶但仍然可以理解,因为在3中Al2修饰的B6环尽管被扭曲,但确是以七配位方式与中心硼原子成键 (图7(b)),这一成键特征与分子轮B82-团簇类似。
4 结论
我们对一系列二元B—Al团簇B7Aln0/-(n=2, 3)进行了量子化学理论研究,旨在理解Al/B取代如何改变硼团簇的结构和成键性质,以及元素B和Al复合对相关体系芳香性的影响。我们发现B7Aln0/-(n=2, 3)团簇的全局极小结构与B8Al-/B9Al-和B9-/B10-不同。团簇B7Al2、B7Al2-和B7Al3都含七边形B7环和垂直于环平面的Al2单元,而B7Al3-则为六配位B7盘状结构 (包括一个面上和两个面内桥连铝原子)。化学键分析表明所有B7Aln0/-(n=2, 3)团簇都具有双重π/σ芳香性,其6π和6σ电子计数符合(4n+2) Hückel规则。在B7Aln0/-(n=2, 3)团簇中铝原子倾向于保留一定数量的定域电子或孤对,这表明二元B—Al复合团簇中的所谓等价Al/B取代并不是完全等价的。此外通过比较两种中性团簇的电子亲和能可理解B7Al3/B7Al3-结构不同的原因。二元B—Al团簇中非完全等价的Al/B取代将为设计新颖硼基团簇和探索独特化学键提供指导。
猜你喜欢
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09
空间科学学报(2020年6期)2020-07-21
空间科学学报(2020年6期)2020-01-08
环球时报(2019-12-05)2019-12-05
当代陕西(2019年6期)2019-04-17
外语学刊(2014年3期)2014-12-03
太空探索(2014年4期)2014-07-19
