
控制高粱分蘖与主茎株高一致性的基因定位
2019-05-31 06:45詹鹏杰于纪珍楚建强平俊爱张福耀
作物学报 2019年6期
王 瑞 凌 亮 詹鹏杰 于纪珍 楚建强 平俊爱,*张福耀,*
1山西省农业科学院高粱研究所 / 高粱遗传与种质创新山西省重点实验室, 山西榆次 030600; 2山西省农业科学院食用菌研究所, 山西太原030031
高粱[Sorghum bicolor(L.) Moench]是全球农业生态系统中重要的粮食和饲料作物。全球年种植面积 4000多万公顷, 总产约 6000万吨。高粱抗旱且耐盐碱和瘠薄土壤, 具有在恶劣环境下生长的能力,被视为干旱和盐碱土壤农业区农业可持续发展的一种重要作物[1-3]。随着对淡水资源需求的增加、边际农田利用的增多和全球气候变暖, 耐干旱、盐碱作物——高粱将在全球粮食需求不断增加的今天显的越来越重要。高粱也是我国的重要旱粮作物之一,全国年种植面积约80万公顷。目前, 我国的农业生产进入新的转型期, 劳动力成本逐年提高, 农民对机械化栽培高粱的渴求与日俱增, 但推广的高粱品种基本不适宜机械化栽培, 一是株高本身偏高, 不适宜机械化作业, 二是分蘖高度与主茎不一致, 高粱穗不在一个层面, 造成收获时丢损严重, 极大地制约了高粱机械化生产的发展[4-5]。因此, 高粱分蘖和主茎株高一致的研究, 对指导高粱育种和选育适宜机械化生产高粱品种具有十分重要的理论意义和应用价值。
国内外对机械化生产高粱品种的研究主要集中在株高和分蘖力这2个性状。Quinby等[6]研究确定了植株高度由4个基因位点控制, 依此命名为Dwl、Dw2、Dw3和Dw4; Pereira 等[7]以 CK60 和 PI229828为亲本杂交得到 F2群体, 计算出 4个控制株高的QTL; Lin等[8]以杂交F2代群体鉴定出6个控制株高的QTL; Rami等[9]以IS2807分别与种质库中2种高粱杂交, 得到2个RIL群体, 以此为研究对象, 得到3个与株高相关的 QTL; Klein等[10]以 Sureno和RTx430为亲本杂交, 选取125株F2鉴定得到2个控制株高性状的QTL; Upadhyaya等[11]用242份微核心种质与39个标记关联分析, 得出5个标记与熟期或株高关联; Upadhyaya等[12]开发了14,739个SNP标记评估株高, 结果发现, 6个 SNP位点与株高关联;Harris-Shultz等[13]以 F2群体发现了 1个新的株高QTL位于第 3染色体上; 苏舒等[14]以 T70和 P607为亲本, 杂交得到F6代RIL检测到6个控制株高的QTL; 刘娟等[15]利用‘河农 16’与‘千三’的 F2:3遗传作图群体的后代材料建立的F8代的132个重组自交系, 检测到2个与株高相关的QTL。分蘖数表现出数量性状的遗传特点, 受多个数量基因位点和环境因子的共同作用[16]。Feltus等[17]利用BTx623 ×IS3620C的F6-8代作为构图群体, 检测到9个控制高粱分蘖数的 QTL; Shehzad等[18]对来自亚洲和非洲27个国家的代表性高粱品种多样性研究发现7个影响分蘖数的 QTL; Shiringani等[19]利用甜高粱 SS79和粒用高粱M71杂交得到的F6代重组自交系群体为定位群体, 检测到 6个影响高粱分蘖数的 QTL; 董维等[20]以T70 × P607杂交得到的F6代RIL群体构建遗传连锁图, 检测到7个与分蘖数相关的QTL。对高粱分蘖和主茎株高一致的研究尚未见报道。本研究通过杂交、自交等方法构建 F2分离群体, 基于SLAF-seq技术, 利用F2遗传分离群体和集群分离法(BSA)对高粱分蘖与主茎株高一致性状进行关联分析, 定位该基因在染色体上的位置, 以期提高我国适宜机械化高粱品种选育水平以及选择的准确性和科学性。
1 材料与方法
1.1 材料
本实验田间部分在山西省农业科学院高粱研究所试验基地(山西榆次和海南三亚)进行, 选用矮杆多分蘖且分蘖整齐一致的品种 K35-Y5为母本, 中高杆有分蘖且分蘖明显高于主茎的恢复系1383为父本杂交得F1代, 冬季在海南三亚种植F1代, 第 2年夏在山西榆次种植 F2代, 株行距为 25 cm × 50 cm, 每公顷留苗约8万株, 保证每个单株充分表达其性状。开花后调查分蘖与主茎株高的一致性, 分蘖株高与主茎株高同等或株高差≤ 5 cm, 记为株高一致, 分蘖株高与主茎株高差> 5 cm记为不一致。
选取F2分离群体中分蘖与主茎株高一致和分蘖明显高于主茎的植株各45株, 构建分蘖与主茎株高一致与分蘖明显高于主茎的 2个极端性状混池, 连同2个亲本作为定位群体进行关联分析。
1.2 SLAF-seq文库构建
选择已经测序完成的高粱基因组作为参考基因组 ( 下 载 地 址 : ftp://ftp.ensemblgenomes.org/pub/plants/release-25/fasta/sorghum_bicolor/, 组装出的基因组大小738.61 Mb, GC含量为43.93%), 根据基因组大小以及 GC含量等信息作为参考基因组进行酶切预测, 根据选定的最适酶切方案, 对检测合格的各样品基因组DNA分别进行酶切。对得到的酶切片段(SLAF标签)进行3′端加A处理、连接Dual-index[21]测序接头、PCR扩增、纯化、混样、切胶选取目的片段, 文库质检合格后用Illumina HiSeq 2500测序。为评估酶切实验的准确性, 选用水稻(Oryza sativa)[22]作为对照进行测序。
1.3 SNP位点的开发与关联分析
利用Dual-index对测序得到的原始数据进行识别, 得到各个样品的 reads。过滤测序 reads的接头后, 进行测序质量和数据量的评估。通过Control数据评估酶切效率, 以此判断实验过程的准确性和有效性。通过将reads与参考基因组比对, 在亲本和混池中开发 SLAF标签, 寻找在亲本中存在多态性的SLAF标签和有 reads覆盖区域的 SNP。将得到的SNP利用 SNP-index方法[23]和ED方法[24]进行关联分析, 获得与性状紧密关联的位点。
1.4 关联分析方法
1.4.1 SNP-index方法原理 SNP-index是通过混池间的基因型频率差异进行标记关联分析的方法[23],主要是寻找混池之间基因型频率的显著差异。
利用两亲本的 SNP数据, 分别计算两混池的SNP-index, 并通过 ΔSNP-index观测可能与性状分离相关的位点。
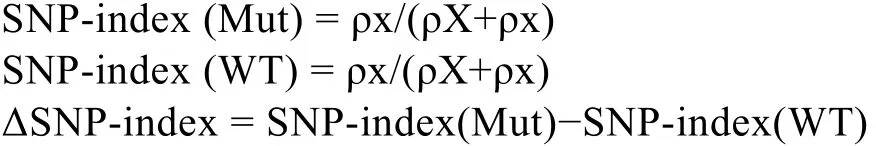
式中, Mut和WT分别为子代的突变池与野生池, ρX和ρx分别为野生型亲本的等位基因, 以及突变型亲本的等位基因在各自池中出现的 read数目。通过ΔSNP-index可以观察每个位点在突变池与野生池之间的差异。
为了消除假阳性的位点, 利用标记在基因组上的位置, 对同一条染色体上标记的ΔSNP-index值进行拟合, 并根据关联阈值, 选择阈值以上的区域作
为与性状相关的区域。
1.4.2 欧氏距离方法原理 欧式距离(Euclidean distance, ED)是利用测序数据寻找混池间存在的显著差异标记, 并以此评估与性状关联区域的方法[24]。ED值越大, 表明该标记在两混池间的差异越大。
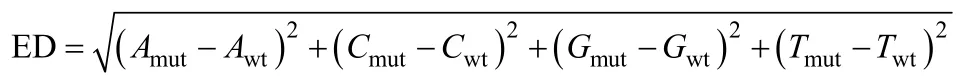
式中,Amut、Cmut、Gmut和Tmut分别为 A、C、G和 T碱基在突变混池中的频率,Awt、Cwt、Gwt和Twt分别为A、C、G和T碱基在野生型混池中的频率。
利用两混池间基因型存在差异的SNP位点, 统计各个碱基在不同混池中的深度, 并计算每个位点ED值, 为消除背景噪音, 对原始 ED值进行乘方处理[24]。
为了消除假阳性的位点, 利用标记在基因组上的位置, 对同一条染色体上标记的ED值进行拟合[24],并根据关联阈值, 选择阈值以上的区域作为与性状相关的区域。
2 结果与分析
2.1 亲本及群体分蘖高度的遗传
K35-Y5是从美国引进的种质, 通过在山西和海南多年种植鉴定, 表现为矮杆多分蘖且分蘖整齐一致, 是适宜机械化生产品种选育的优良亲本系。1383是我国主干高粱杂交种‘晋杂 12号’的恢复系, 中高杆有分蘖且明显高于主茎, 由它配制的晋杂12号在生产中一直表现分蘖高、弱苗高、整齐度差。
选用K35-Y5与1383杂交, F1植株表现为有分蘖且分蘖比主茎高, 说明分蘖与主茎株高一致性状为隐形, 而分蘖高于主茎性状为显性。F2分离群体中分蘖与主茎株高不一致与一致的分离比例为239∶72, 经卡方(χ2)检验, χ2=0.57<χ20.05=3.84, 分离符合 3∶1, 说明分蘖与主茎株高一致性状受一对隐性核基因控制。
2.2 酶切方案与建库评估
对高粱参考基因组序列进行电子酶切预测, 确定限制性内切酶为RsaI +HaeIII, 酶切片段长度在364~414 bp的序列定义为 SLAF标签, 预测可得到103,902个SLAF标签。统计SLAF标签在各染色体上的数量(表1), 绘制SLAF标签在染色体上的分布图(图1)。由图可知, SLAF标签在基因组各染色体上分布基本均匀, 酶切方案可行。
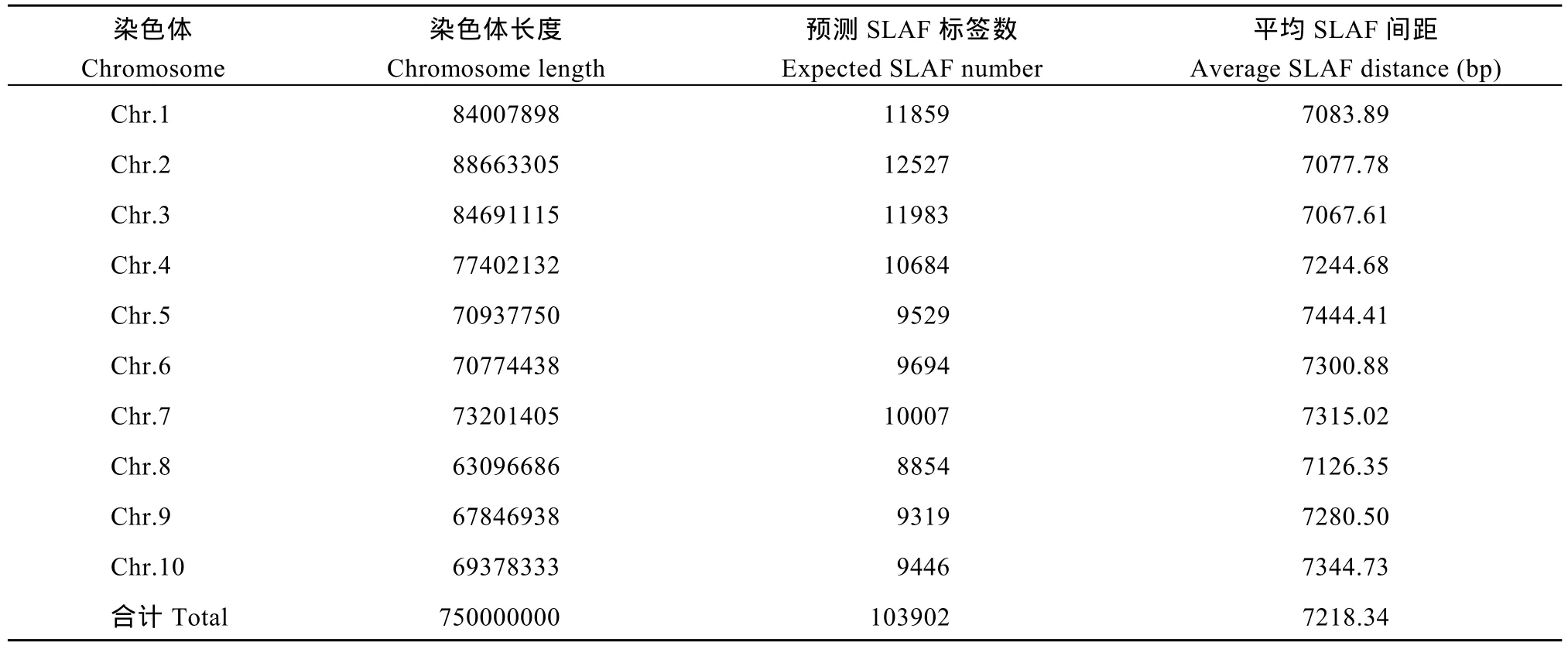
表1 SLAF标签在各染色体上的数量统计Table 1 Number of SLAF labels on each chromosome
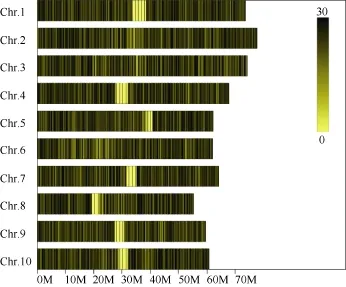
图1 SLAF标签在参考基因组各染色体上的分布Fig. 1 Distribution of SLAF on reference genome
为进一步评估酶切方案的有效性, 以水稻(Oryza sativa)作对照, 其基因组大小为374.31 Mb (下载地址:http://rice.plantbiology.msu.edu), 通过 BWA[25]软件将对照的测序 reads与参考基因组比对(表 2和图 2)显示, 本次实验双端比对效率在 93.35%, 酶切效率为 90.60%, 对照测序 reads插入片段的长度均集中分布在360~410 bp之间, 说明SLAF建库正常。
2.3 测序数据统计与评估
采用100 bp×2作为后续使用的数据, 进行数据评估和分析。测序质量值(Q)是评估高通量测序单碱基错误率的重要指标, 测序质量值越高对应的碱基测序错误率越低。共获得30.80 M reads数据, 测序平均Q30为91.70%, 平均GC含量为45.79% (表3)。用于评估实验建库的准确性的水稻(Control)测序获得0.19 M reads的数据量。
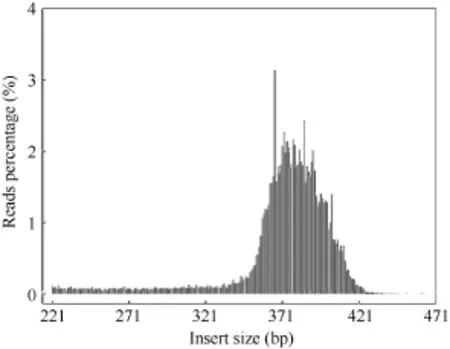
图2 对照reads插入片段分布Fig. 2 Distribution of observed control insert size

表2 水稻测序reads比对分析Table 2 Alignment analysis of obtained reads in Oryza sativa
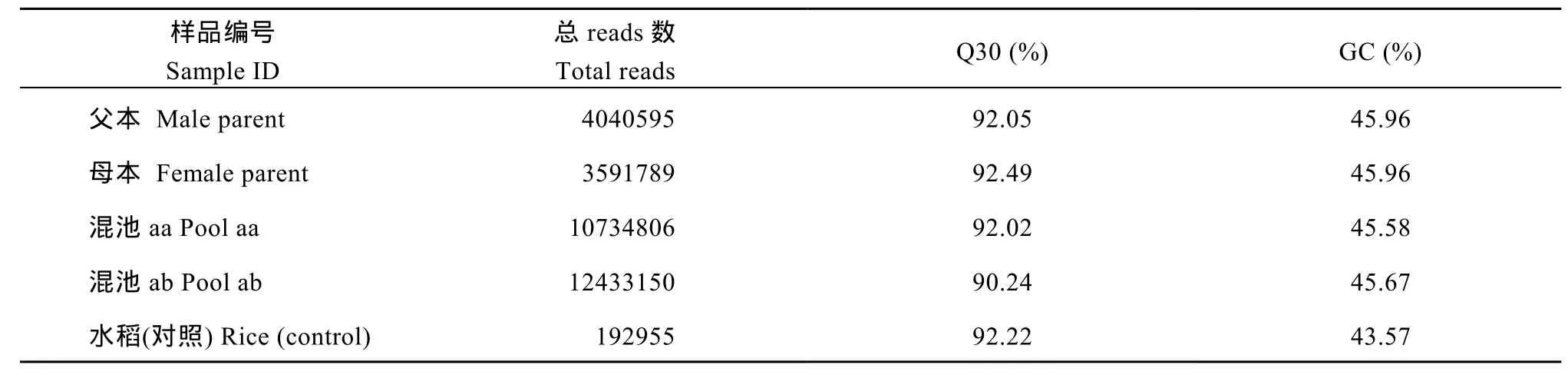
表3 各样品测序数据统计表Table 3 Statistics of sequencing for each sample
2.4 SLAF标记开发
利用参考基因组共开发133,246个SLAF标签,SLAF标签亲本平均测序深度为 22.68×, 混池平均测序深度为 61.94× (表 4); SNP 的检测主要使用GATK软件工具包实现, 所有样品的SNP统计信息见表5。统计不同染色体上的SLAF标签与SNP标记的分布(表 6); 根据 SLAF在染色体上的分布, 绘制SLAF标签和多态性SLAF标签的染色体分布图,由图3可以看出, 开发的SLAF标记分布较均匀。
2.5 关联分析
2.5.1 SNP-index方法关联结果 在 SNP-index关联分析前, 先从319,428个SNP中, 过滤掉有多重突变的SNP位点、混池中read支持度小于4的位点、亲本中不存在的 SNP位点, 最终获得 59,407个SNP。利用 SNP-index方法[23]计算关联值, 并采用SNPNUM方法对ΔSNP-index进行拟合, 取每个SNP附近200个SNP的ΔSNP-index的中值作为该位点拟合后的关联值。两个混池分别的 SNP-index及ΔSNP-index的分布如图4所示:
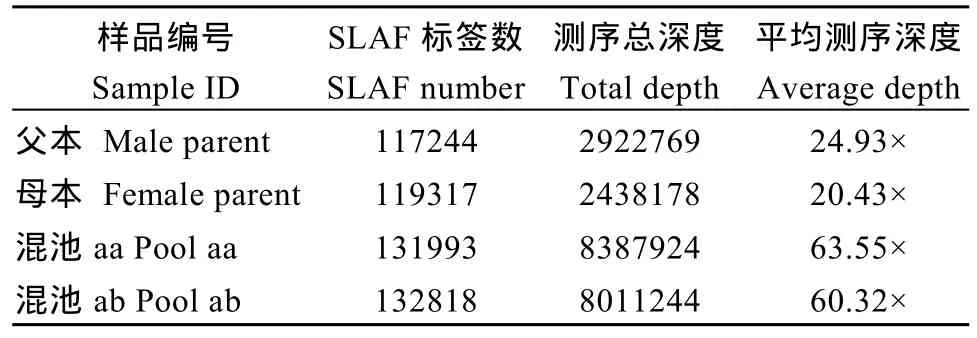
表4 SLAF标签统计Table 4 Statistics of SLAF
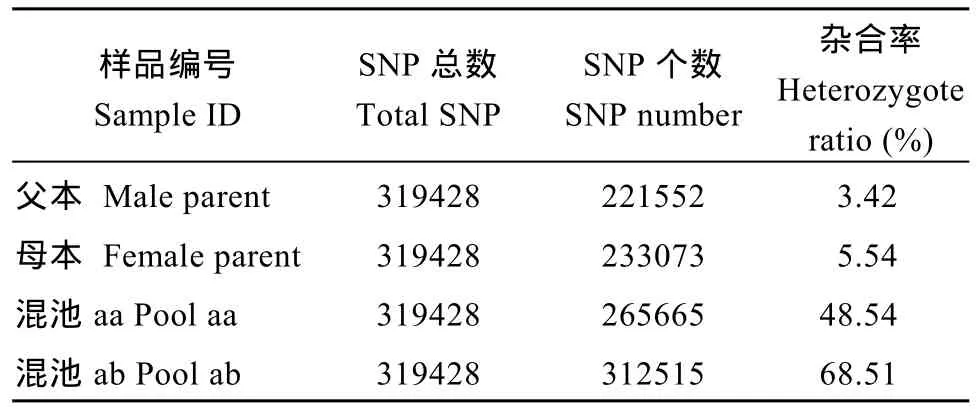
表5 SNP信息统计Table 5 Statistics of SNP information
根据计算机模拟实验[26]计算结果, 当置信度为0.90时, 定位区域在第 9染色体上的 54,788,026~56,740,873 (1.95 M)区间内(表 7)。
2.5.2 ED方法关联结果 在ED法关联分析前,先从319,428个SNP中过滤任一混池中read支持度小于 4的位点 212,186个, 得到高质量的可信SNP位点共107,242个, 并在此基础上识别两混池间差异的位点共100,261个。利用ED方法[24]计算关联值, 并取原始ED的3次方作为关联值以达到消除背景噪音的功能, 然后采用局部线性回归LOESS方法对 ED值进行拟合, 关联值分布见图 5。
取所有位点拟合值的 median+3SD作为分析的关联阈值[24], 计算得 0.0457, 根据关联阈值判定,定位区域在第 9染色体的 30,964,929~38,410,608(7.45 M)和 43,824,552~59,633,348 (15.81 M)区间内(表 8)。
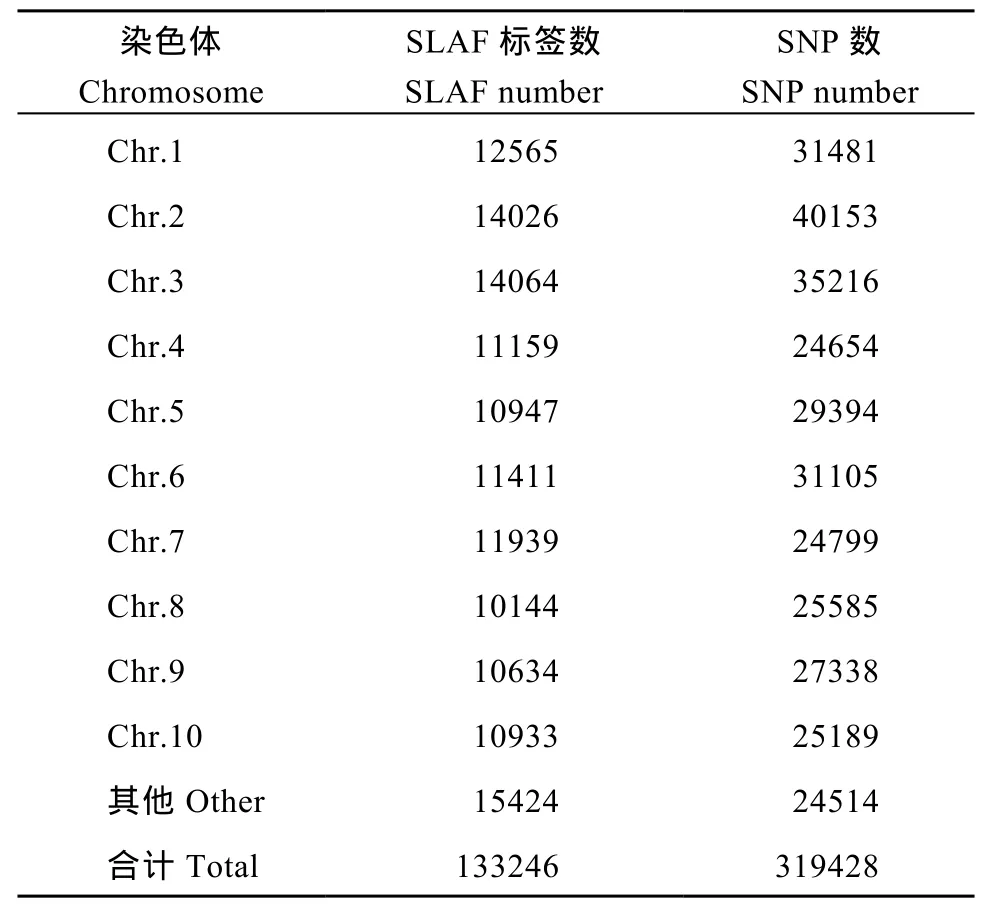
表6 SLAF标签和SNP标记在染色体上的分布统计Table 6 Distribution statistics of SLAF and SNP on genome
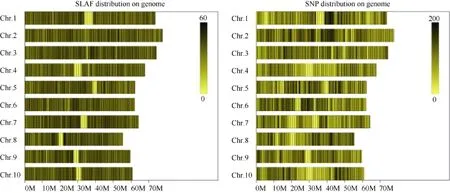
图3 SLAF标签和SNP标记在染色体上的分布Fig. 3 Distribution of SLAF and SNP on genome
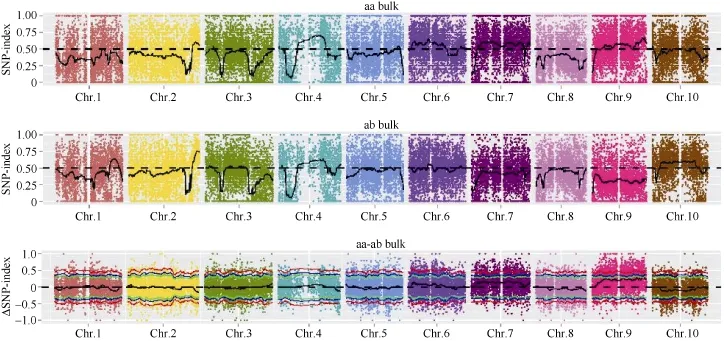
图4 SNP-index关联值在染色体上的分布Fig. 4 Distribution of SNP-index associated values on genome
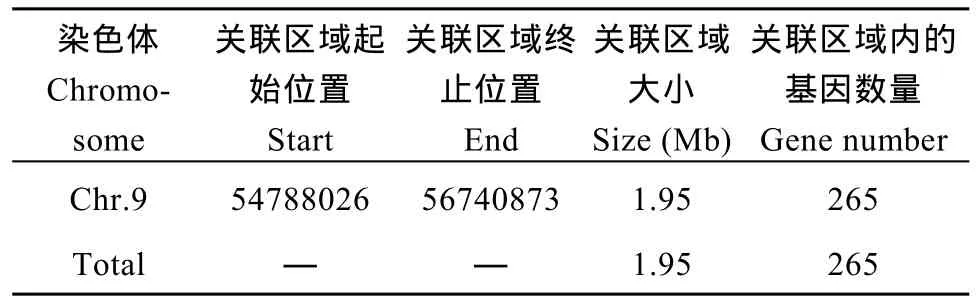
表7 关联区域信息统计表Table 7 Statistics of associated region information
2.5.3 候选关联区域筛选与功能分析 对这2种方法取交集, 以期得到更准确的结果, 得到的交集是定位区域在第 9染色体上的 54,788,026~56,740,873区间内, 该关联区域长度1.95 Mb, 在关联区域内共有 265个候选基因。针对关联区域内的基因, 分析在外显子区域两个亲本之间差异的 SNP信息, 对 SNP进行变异的注释(表 9), 共发现存在非同义突变的 SNP4个, 对应到Sobic.009G197901.1、Sobic.009G213300.1和Sobic.009G221200.1三个基因,初步确定这些基因是与性状直接相关的功能基因。
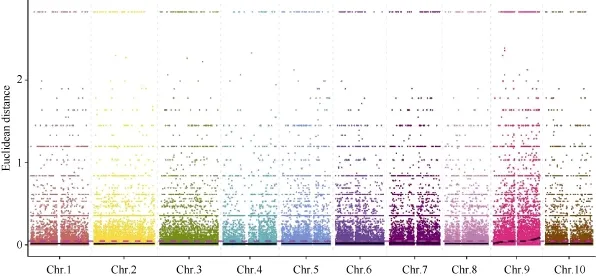
图5 ED关联值在染色体上的分布Fig. 5 Distribution of ED associated values on genome
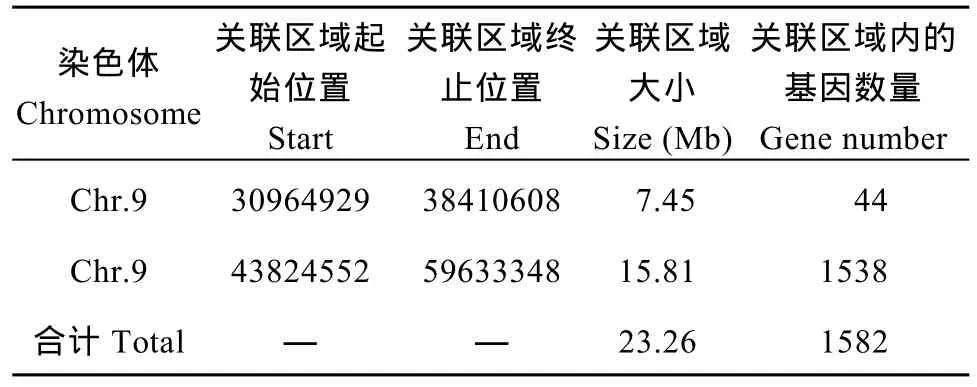
表8 关联区域信息统计表Table 8 Statistics of associated region information
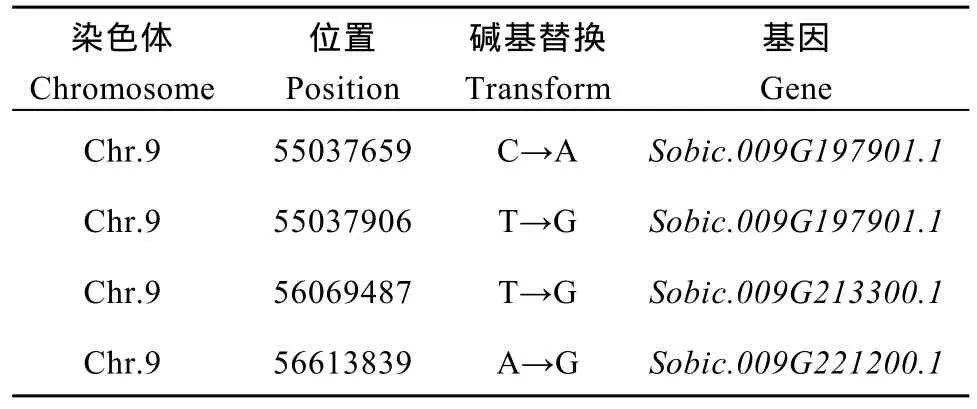
表9 SNP标记信息Table 9 Information of SNP
2.6 相关连锁分子标记的开发及验证
在4个非同义突变的SNP两端设计引物(表10),对双亲的基因组 DNA进行 PCR扩增, 用琼脂糖凝胶电泳检测扩增产物。将扩增产物回收, 测序; 对测序结果进行多序列对比, 获得基因序列特异差异(图6)。分蘖与主茎株高不一致亲本1383和分蘖与主茎株高一致亲本K35-Y5在第55,037,659 bp处分别为A和C; 在第 55,037,906 bp处分别为 G和 T; 在第56,069,487 bp处分别为G和T; 在第56,613,839 bp处分别为G和A。说明这4个SNP是与分蘖与主茎株高一致性状直接相关的。
3 讨论
对高粱分蘖性已有广泛研究, 影响分蘖性的主要因素有水肥条件、温度、密度等[27]。水肥条件充分, 分蘖期早且长, 分蘖早且多; 不同品种分蘖性具有随播种至分蘖期土壤温度的升高而降低的趋势,温度高, 生长快, 迅速进入拔节期, 缩短分蘖时期;在北方地区, 随着种植密度的增加单株分蘖数明显减少, 高粱分蘖性受环境影响较大。但高粱分蘖与主茎的高度差这一性状基本不受环境的影响, 分蘖与主茎高度一致这一性状遗传稳定, 已在适宜高粱机械化育种中应用, 育成一批适宜机械化生产的高粱品种[28]。Upadhyaya等[11]用 242份微核心种质与39个标记关联分析, 得出5个标记与熟期或株高关联, 分别位于第 6、第 9、第 10染色体上。Brown等[29]用包含119个单株的RIL群体鉴定得到与株高相关的4个QTL, 分别位于第3、第6、第7、第9染色体上。Shiringani等[30]建立了包含188个单株的RIL群体, 利用 157对引物, 得到 5个关于株高的QTL, 分别位于第1、第2、第5、第6、第9染色体上。Feltus等[17]利用 BTx623×IS3620C 的 F6-8代作为构图群体, 共检测到 9个控制高粱分蘖数的 QTL,多数集中于第1和第6染色体上, 第5、第7和第9染色体上各检测到1个QTL。不同的研究者研究结果不尽相同, 但大多研究结果都关联到第9染色体。本研究对高粱分蘖与主茎株高一致性基因的定位与前人研究结果一致, 所定位到的基因也位于高粱第9染色体上, 说明高粱第9染色体不仅存在分蘖性状的基因, 而且还有分蘖与主茎株高一致性的基因。

表10 引物序列和信息Table 10 Sequence and information of the primers

图6 引物SNP1(A)、SNP2(B)、SNP3(C)、SNP4(D)对双亲的测序结果Fig. 6 Sequencing results of the parents with primer SNP1(A), SNP2(B), SNP3(C), and SNP4(D)
基于 SLAF-seq技术的基因定位, 是集 BSA技术、生物信息学和高通量测序技术为一体的一种简单高效的基因定位方法。目前, 利用此类方法定位的有番茄雄性不育基因[31], 甘蓝型油菜抗霜霉病基因[32], 油菜与种子重量高度相关的基因[33]等。本研究利用SLAF-seq技术, 将高粱分蘖与主茎株高一致基因定位于第 9染色体上的 54,788,026~56,740,873区间内, 关联区域长度1.95 Mb。
4 结论
分蘖与主茎株高一致性状由1对隐性核基因控制。利用SNP-index法和Euclidean distance法及取两者交集进行关联分析, 得到一个关联区域, 位于第 9染色体上的 54,788,026~56,740,873区间内, 关联区域长度1.95 Mb。在该关联区域内发现4个非同义突变的 SNP, 它们和分蘖与主茎株高一致性状相关。对应到Sobic.009G197901.1、Sobic.009G213300.1
和Sobic.009G221200.1三个基因上, 这些基因可能是与性状直接相关的功能基因。通过进一步精细定位, 可以确定该目标基因。
猜你喜欢
安徽农业科学(2022年19期)2022-10-29
安徽农业大学学报(2022年3期)2022-10-25
古今农业(2022年2期)2022-08-15
青年文学家(2022年1期)2022-03-11
今日农业(2021年20期)2021-11-26
种子(2021年7期)2021-08-19
农村百事通(2019年17期)2019-10-08
中国烟草科学(2019年1期)2019-09-10
现代农业科技(2017年1期)2017-03-06
江苏农业科学(2016年8期)2017-02-15
