
基于线粒体COⅠ序列的南海北部栉江珧遗传多样性分析
2019-05-29 06:58于非非钟智明牛素芳杜晓东许开航林子腾张颖懿王家晋刘漫玲
水生生物学报 2019年3期
于非非 钟智明 牛素芳 杜晓东 许开航 林子腾 张颖懿 王家晋 刘漫玲
(广东海洋大学水产学院, 湛江 524088)
栉江珧(Atrina pectinata), 隶属于软体动物门(Mollusca), 瓣鳃纲(Lamellibranchia), 贻贝目(Mytiloida), 江珧科(Pinnidae), 江珧属(Atrina)[1], 有带子、大海红、海锨、大海荞麦、土杯、马蹄等俗称。栉江珧为一种广温、广盐的稳居性贝类, 广泛分布于温、热带泥沙质近海地区, 我国的渤海、黄海、东海和南海四大海域均有分布, 其中南海是我国栉江珧分布种类最多、产量最高的海区[1]。栉江珧闭壳肌肥大、肉质鲜嫩, 具有较高的经济价值和药用价值[2], 在我国南海沿岸地区备受欢迎。然而, 栉江珧的人工养殖技术尚不完善, 一般采用半人工养殖方法, 即采捕野生栉江珧苗种、然后选择合适海区进行集中养殖的方式[3]。多年的过度捕捞和人工养殖的相对滞后使栉江珧的野生资源遭到大量破坏, 种质资源明显退化[4]。这与南海沿岸日益上升的需求量相违背, 对南海栉江珧资源进行调查和保护成为亟待解决的科学问题。
细胞色素氧化酶亚单位Ⅰ(Cytochrome Oxidase Ⅰ,COⅠ)是一种线粒体DNA标记, 因其结构简单、进化速度快、母系遗传、易于扩增等特点,被广泛应用于不同生物群体的遗传多样性分析[5]。近年来有研究者基于COⅠ序列对不同地理位置栉江珧的群体遗传性进行了分析, 如严加坤等[4]调查了长岛、日照、文登、湛江、海口五个城市的野生栉江珧资源, 发现南方群体和北方群体之间存在极大遗传分化, 而南北方群体内部几乎没有遗传分化。Xue等[6]利用COⅠ基因分析了辽宁獐子岛、山东蓬莱、山东刘公岛、山东荣成、山东红岛、山东日照、江苏连云港、浙江舟山、福建福州和韩国海州(Haeju)8个地区的栉江珧资源, 发现来自中国北方海域的7个群体虽然遗传多样性较高, 但没有遗传分化; 而中国和韩国样品间存在明显的遗传分化。以上利用COⅠ基因对栉江珧的研究多聚焦在中国北部海域, 或者更关注南北方海域群体的差异, 而对于栉江珧分布种类最多、产量最高的南海北部海域群体内部的资源调查研究较少。
本研究基于COⅠ基因序列对我国南海北部7个沿海城市的栉江珧群体进行群体遗传学研究,从遗传多样性、遗传分化和历史动态探讨南海北部栉江珧的系统地理格局, 以期阐明南海北部栉江珧的群体遗传特征, 评估其遗传资源状况, 为资源保护、人工养殖和良种选育提供理论储备。
1 材料与方法
1.1 样品采集
采集了汕头(ST)、阳江(YJ)、湛江(ZJ)、海口(HK)、琼海(QH)、北海(BH)、防城港(FCG)近海等7个海区野生群体, 共191只栉江珧个体(图1和表1)。对所有样品进行形态学鉴定后取其闭壳肌,-80℃保存备用。
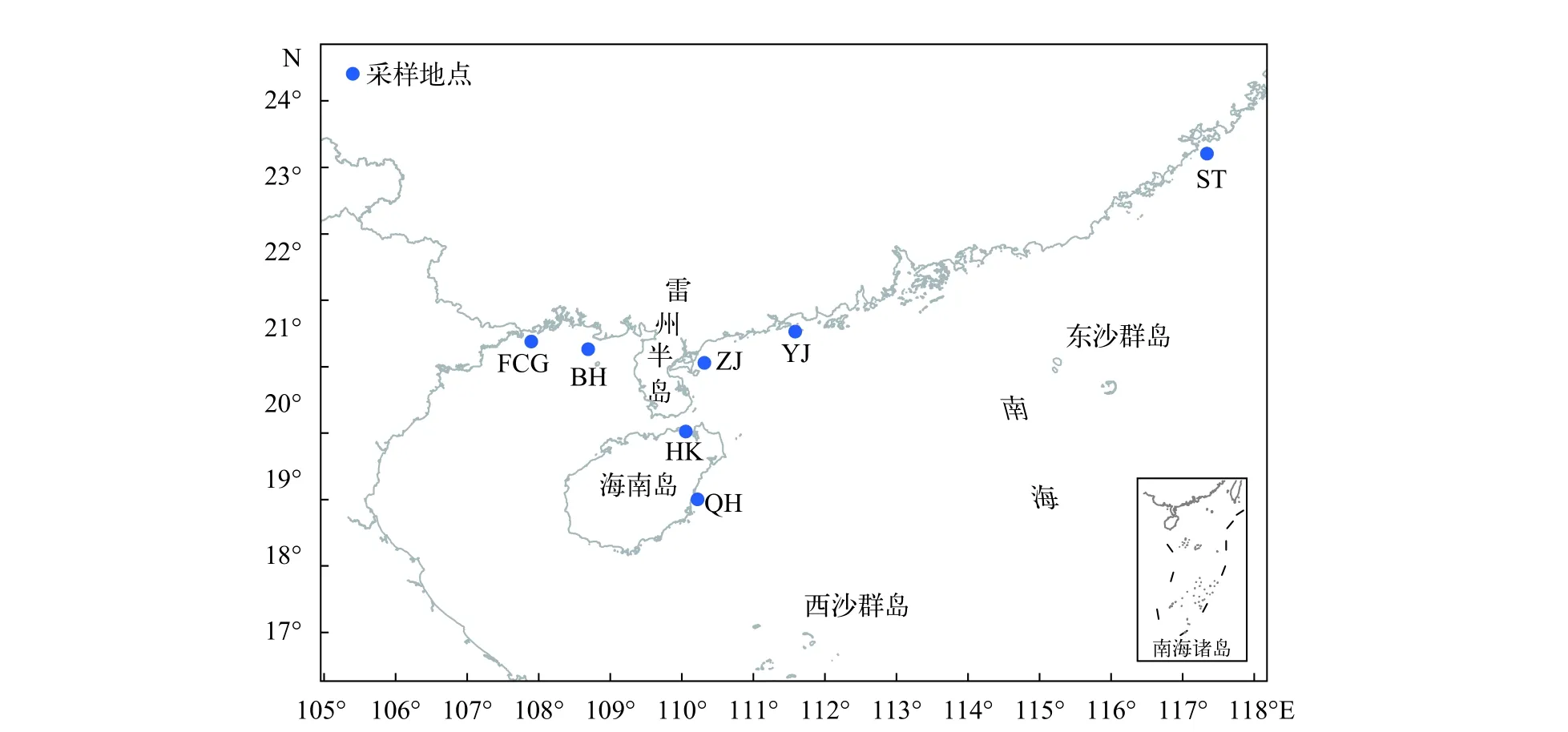
图1 南海北部栉江珧7个群体的采样地点Fig. 1 Sample locations for seven A. pectinata populations from the Northern South China Sea
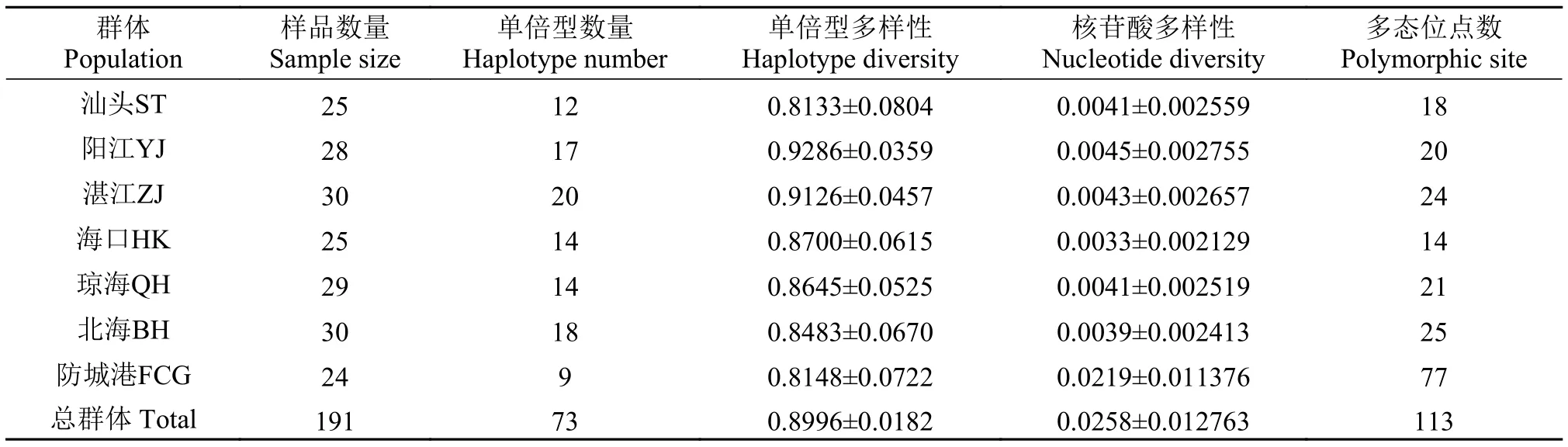
表1 南海北部栉江珧7个群体的采样信息和遗传多样性参数Tab. 1 The sampling information and genetic diversity indices for 7 A. pectinata populations from the Northern South China Sea
1.2 DNA提取和目的基因的扩增
用E.Z.N.A.R Tissue DNA试剂盒(OMEGA)提取各样品基因组DNA, 1%琼脂糖凝胶电泳检测DNA的纯度和浓度。COⅠ基因片段扩增所用引物序列为: LCO1490 (GGTCAACAAATCATAAA GATATTGG)和HCO2198 (TAAACTTCAGGGT GACCAAAAAATCA)。PCR反应体系为25 μL 2×EasyTaqPCR SuperMix (TransGen)、正反引物(10 μmol/L)各1 μL, 基因组DNA (100 ng/μL) 2 μL,ddH2O补齐50 μL。PCR反应程序为: 94℃预变性3min; 94℃变性30s, 57℃退火30s, 72℃延伸1min,35个循环; 最后72℃延伸10min。PCR产物经1%琼脂糖凝胶电泳检测后由生工测序。
1.3 数据处理
利用DNAStar 7.1和Clustal X 1.81[7]进行序列校对和编辑; 利用DnaSP 5.10.01[8]计算单倍型数目、单核苷酸变异位点数和多态简约信息位点数;利用Arlequin 3.5.1.2[9]统计序列的平均碱基组成, 计算单倍型多样性(h)和核苷酸多样性(π)等群体遗传学参数; 利用MEGA 6.06[10]计算转换百分比、颠换百分比和转换/颠换比率(Ts/Tv), 选择DNA序列的最佳替换模型。利用MrBayes 3.2.6构建单倍型系统进化树, 利用Network5.0.0.1(http://www.fluxus-engineering.com/share-net.htm)中的中介连接网络法构建单倍型网络支系图[11]
群体遗传结构采用3种方法进行分析: (1)采用分化固定指数Fst评价两两群体间的遗传差异, 检验结果的显著性; (2)根据样品来源、遗传学参数和Fst, 将7个栉江珧群体划分为2个组: 汕头(ST)、阳江(YJ)、湛江(ZJ)、海口(HK)、琼海(QH)和北海(BH)6个栉江珧群体为一组(L1组), 防城港群体(FCG)单独为一组(L2组), 通过AMOVA分析估算遗传变异在组群间、组群内群体间和群体内的分布,进行显著性分析; (3)通过Exact检验检测单倍型在群体间分布频率的差异。
群体历史动态采用2种方法进行检测: (1)采用Tajima'sD检验和Fu'sFs检验检测中性假说是否成立, 并进行显著性分析; (2)基于核苷酸不配对分布检验南海北部栉江珧群体是否存在数量和空间扩张, 并通过拟合优度检验(Goodness of fit test)检测核苷酸不配对的观测分布和预期分布之间的一致性, 并估算群体历史扩张时间。在本研究中,COⅠ基因的分化速率采用2.4%/MY (Million year, MY)[6],世代时间设置为1年[12]。
2 结果
2.1 序列分析
本研究共获得7个群体191条长度为600 bp的COⅠ基因部分序列, 其G、C、A、T的平均含量分别为24.05%、14.81%、19.18%和41.96%。共检测到113个核苷酸变异位点(18.8%), 包括14个单核苷酸变异位点, 99个多态简约信息位点, 无插入和缺失位点。转换数共有107次, 明显高于颠换数(19次),Ts/Tv=15.33。转换和颠换在G、C、A、T之间均有发生, 其中C-T (53.68%)转换多于G-A(40.88%)转换, T-G (1.79%)和T-A (1.66%)颠换稍多于C-G (1.05%)和C-A (0.92%)颠换。突变位点均匀地分布在COⅠ基因上, 但在群体间的分布差异较大, 77个多态位点来源于FCG群体。
2.2 单倍型和遗传多样性分析
COⅠ单倍型在7个群体中的分布见表1和表2。113个核苷酸变异位点共定义73个单倍型(Gen-Bank登录号为MH536017—MH536089), 包括58个独有单倍型和15个共享单倍型。在共享单倍型中,3个单倍型(H23、H36和H44)为6个群体所共有,1个单倍型(H2)为5个群体所共享, 1个单倍型(H29)为3个群体所共享, 10个单倍型(H1、H18、H20、H34、H47、H51、H52、H54、H61和H62)为2个群体所共享。H44出现的频率最高(占总样品的31.41%), 这表明这个单倍型是南海北部栉江珧在长期进化过程中形成的较为稳定的优势基因型。值得注意的是, 防城港群体具有9个单倍型(H65-H73), 均为独有单倍型。
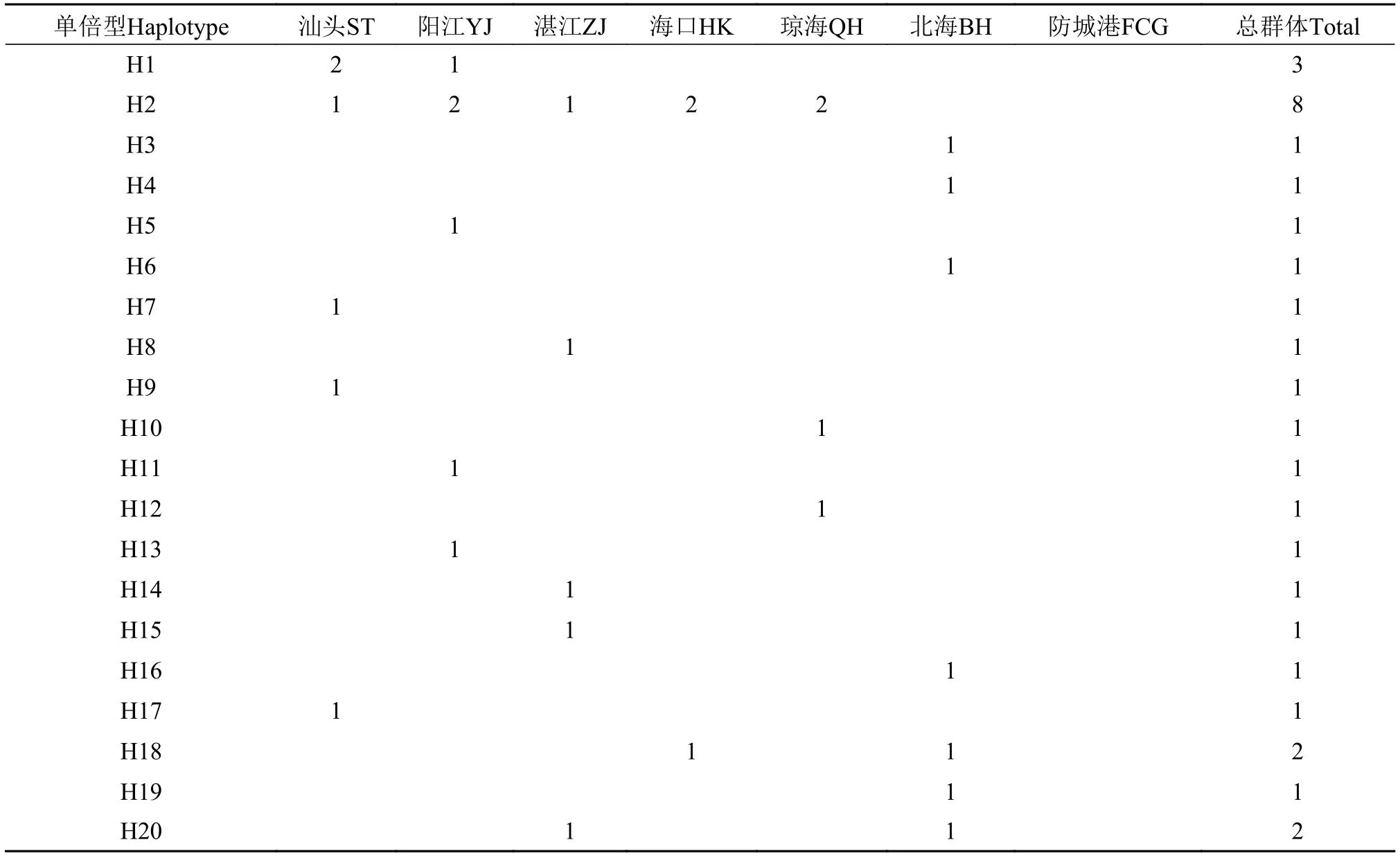
表2 南海北部栉江珧7个群体的单倍型分布情况Tab. 2 Haplotype distribution of 7 A. pectinata populations from the Northern South China Sea

续表2
遗传多样性参数如表1所示, 南海北部栉江珧总群体的单倍型多样性和核苷酸多样性分别为0.8996和0.0257, 表明南海北部栉江珧群体的遗传多样性目前仍处于较高的水平。在7个群体中,L1组6个群体的单倍型多样性为0.8133—0.9286, 核苷酸多样性为0.0033—0.0045, 表现出高h低π遗传多样性模式; 而防城港群体的单倍型多样性为0.8148, 核苷酸多样性较高(0.0219), 表现出高h高π遗传多样性模式。
2.3 群体遗传学关系分析
以旗江珧(Atrina vexillum)COⅠ基因序列为外群, 构建栉江珧73个单倍型的贝叶斯推理树(图2)。结果显示, L1组6个群体的各单倍型交叉出现在系统发育树中, 并未表现出与地理位置相对应的谱系结构; L2组的独有单倍型(H65-H73)聚为一个独立的进化分支, 显示了较远的遗传距离。单倍型网络支系图(图3)具有1个主体单倍型(H44), 该主体单倍型在6个群体中均有分布, 且出现频率较高,说明它可能是起源于母系祖先的主体单倍型; 其他单倍型呈辐射状分布在主体单倍型周围, 其中L2组的9个独有单倍型作为一个独立分支, 与其他单倍型具有较远的遗传距离。这暗示着南海北部栉江珧北部湾的防城港群体与其他群体间存在显著的遗传结构。
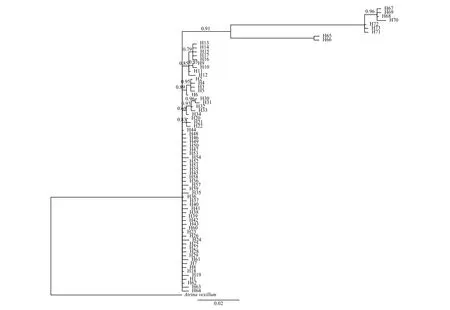
图2 南海北部栉江珧COⅠ单倍型贝叶斯推理树Fig. 2 Bayesian inference tree for A. pectinata from the Northern South China Sea based on COⅠ
2.4 群体遗传结构
由表3可知, L1组6个群体间的Fst值均较小(-0.0200— -0.0055), 且统计学差异不显著(P>0.05);但L 2组与L 1组6个群体间的Fst值均较大(0.8729—0.8821), 且统计学差异极显著(P<0.01)。分子方差分析(Analysis of molecular variance, AMOVA)结果显示(表4), 组群间、组群内群体间和群体内个体间的遗传变异比例分别为97.01%、-0.1%和3.09%, 组群间、组群内群体间和群体内个体间的固定系数Fct、Fsc和Fst分别为0.9701 (P>0.05)、-0.0324 (P>0.05)和0.9691 (P<0.01)。Fst值和AMOVA结果表明, L1组6个群体间在分子水平不存在明显的遗传分化, 但L2组与L1组6个群体间存在显著遗传分化。Exact检验结果显示, 单倍型在L1组6个群体间的分布频率并无显著差异, 但在L2组与L1组间差异显著。

图3 南海北部栉江珧COⅠ单倍型支系图Fig. 3 Haplotype network of A. pectinata from the Northern South China Sea based on COⅠ

表3 南海北部栉江珧7个群体间的遗传分化指数FstTab. 3 The pairwise Fst among seven populations of A. pectinata from the Northern South China Sea
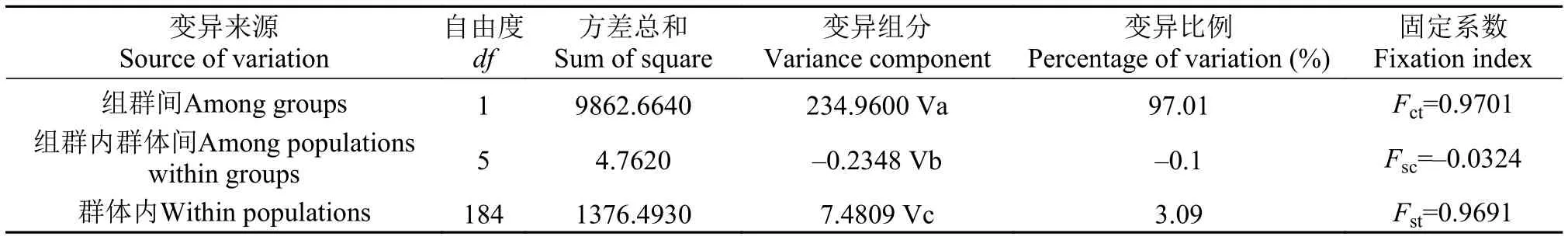
表4 南海北部栉江珧7个群体间的AMOVA分析Tab. 4 Analysis of molecular variance (AMOVA) among seven populations of A. pectinata from the Northern South China Sea
2.5 群体历史动态
由于FCG群体与其他6个群体间具有显著的遗传分化, 故将FCG群体单独进行历史动态分析。中性检验结果显示, FCG群体所在L2组的Tajima'sD检验(D= -1.4320,P=0.0565)为不显著负值, Fu'sFs检验(Fs=4.9540,P=0.9620)为不显著正值, 说明FCG群体进化符合中性假说。L1组6个群体间无显著的遗传分化, 故将其合并为一个大群体进行历史动态分析。结果显示, Tajima'sD检验(D= -2.3190,P=0)和Fu'sFs检验(Fs= -26.8316,P=0)均为显著负值。基于数量扩张模型和空间扩张模型的核苷酸不配对分布均呈单峰分布(图4), 且观测分布和期望分布拟合优度较好(SSD),Hri值小且不显著(表5)。中性检验和核苷酸不配对分布结果均提示南海北部栉江珧L1组6个群体在历史上可能发生过数量和栖息地扩张事件。数量扩张模型和空间扩张模型下的τ值分别为1.2830和1.0880 (表5), 据此推算南海北部栉江珧群体扩张时间约在75500—89000年前。

图4 南海北部栉江珧总群体COⅠ核苷酸不配对分布图Fig. 4 Mismatch distributions of A. pectinata from the Northern South China Sea based on COⅠ

表5 南海北部栉江珧总群体COⅠ核苷酸不配对分布参数Tab. 5 Mismatch distribution indices of A. pectinata from the Northern South China Sea based on COⅠ
3 讨论
3.1 群体遗传多样性
单倍型多样性(h)和核苷酸多样性(π)是衡量物种遗传多样性的2个重要指标[13]。在本研究中, 南海北部栉江珧L1组群呈现出较高的单倍型多样性(0.8133—0.9286)和较低的核苷酸多样性(0.0033—0.0045), 即高h低π模式。这种高h低π的遗传参数特征通常是由一个较小的有效群体经过近期快速扩张成一个大的群体所引起的[14]。在末次冰盛期, 南海海平面下降约100—120 m, 变成一个半封闭的内陆海(图1), 成为众多海洋生物的冰期避难所[15,16]。在冰期恶劣的环境下, 南海栉江珧的分布范围和数量可能经历了一定程度的收缩; 随着间冰期气温回升, 海平面上升, 产生大量近岸栖息地, 冰期避难所内残留的栉江珧可能发生快速扩张重新进入南海大陆架。此过程南海栉江珧群体积累了单倍型的多样性, 但尚缺乏足够的时间积累核苷酸序列的多样性[11]。而防城港群体显示了较高的单倍型多样性(0.8148)和核苷酸多样性(0.0219), 这暗示着北部湾丰富的生态资源和相对稳定的生存环境对栉江珧保持遗传多样性产生了重要影响。
单倍型多样性和核苷酸多样性分析结果表明,南海北部栉江珧群体的遗传多样性目前仍处于较高的水平, 这可能与栉江珧广阔的生境范围有关。栉江珧在我国东海、渤海、黄海和南海海域均有分布, 广阔的生境范围使其面临较小的自然选择压力, 积累较多的遗传变异。虽然自第四纪以来, 全球温度的大幅度升降使许多物种的遗传多样性损失严重[17], 但栉江珧作为一种底栖动物可能受到的影响较小, 保留了较高的遗传多样性。
3.2 群体遗传结构
在本研究中, AMOVA分析、Fst值、单倍型系统发育树和网络支系图结果均表明, L1组6个栉江珧群体的遗传差异不显著, 不存在显著的群体遗传结构。造成L1组6个群体遗传分化不显著的原因可能有三点: 一是扩散生活史特征。对于有幼虫阶段的海洋生物, 浮游期(Pelagic larval duration, PLD)对于基因交流和遗传结构具有重要影响[18,19]。栉江珧的浮游期约30d左右, 比栉孔扇贝(Chlamys farreri)(约15d)、中国蛤蜊(Mactra chinensis)(约10d)等双壳类具有更长的浮游期[18], 这暗示着栉江珧具有较强的扩散能力, 可以越过地理环境和水深条件,产生更多的基因交流。二是洋流的输送作用。南海沿岸栉江珧5月份进入成熟期和排放期, 排放期可一直延续到12月份, 在此期间栉江珧的性腺分批多次排放[20]。西南季风漂流、南海暖流、粤东沿岸流和海南岛以东沿岸流等北向海流[21,22]有利于QH、ZJ等地方群体卵和幼虫的的北上扩散; 而东北季风漂流、广东沿岸流和黑潮南海分支等南向海流[21,22]则可促进ST群体向南迁移, 从而进行基因交流。三是目前一般认为海洋生物是从海洋到陆地的迁移方式, 陆地周围的淡水、浊流和人类活动对生物体的扩散形成一个地理和生态的障碍[23]。但栉江珧适应能力强, 从潮下带到水下100 m、泥质或砂质海区皆可分布[24,25], 这意味着栉江珧比其他贝类具有更强的克服地理和生态障碍的能力, 能够被洋流输送更远的距离, 产生更多的基因交流。
值得一提的是防城港群体与其他6个群体间产生了明显的遗传分化, 具有显著的遗传结构, 有3个可能的解释: 一是北部湾是一个半封闭的海域, 位于大陆架边缘, 于古近系断陷期形成, 南海栉江珧很可能在此时期发生了分化[26], 其中L1组群随后进行了大规模扩散, 而防城港群体由于地理位置所限,扩散程度则相对较小, 经过多年积累形成了遗传分化。二是基于生态竞争不相容原理。当两个相近的物种或个体共存于一个相对狭小的生境中时, 他们可能会划分生存空间和食物资源, 以最大程度地减少生态位重叠和竞争[27,28]。长期空间和食物的差异导致两个物种最终产生遗传分化, 尤其体现在防城港群体和北海群体间。三是北部湾的自然资源对于栉江珧群体而言非常丰富, 尤其是红河的汇入带来了丰富的海底沉积物。北部湾的红树林资源亦很丰富, 空间异质性强, 可以作为双壳类幼虫的天然屏障, 不但给幼虫提供了多样性的食物和小生境, 也减少其他物种对幼虫的掠夺[26,29], 从而使北部湾狭小空间内产生多样性的系统分化成为可能。
3.3 资源管理
目前, 南海北部栉江珧种质资源明显退化[4], 而物种的遗传多样性是生存和进化的前提, 丰富的遗传多样性意味着较大的进化潜力, 也是生物适应环境变化的必要条件[30]。本研究表明, 北部湾防城港栉江珧群体COⅠ的遗传多样性水平较高; 汕头、阳江、湛江、海口、琼海和北海6个群体的遗传多样性相对偏低, 其中海口群体的遗传多样性最低。为避免栉江珧种质资源严重衰退的现象, 应当加强栉江珧的管理和保护, 尤其是面临较大捕捞压力的南海北部沿岸群体。
猜你喜欢
广西电业(2022年7期)2022-10-13
世界科学技术-中医药现代化(2022年3期)2022-08-22
肝博士(2022年3期)2022-06-30
小猕猴智力画刊(2022年4期)2022-05-23
黄河之声(2021年10期)2021-09-18
中国设备工程(2020年7期)2020-06-28
中国饲料(2019年19期)2019-03-25
中国-东盟博览(旅游版)(2018年7期)2018-05-14
军营文化天地(2017年1期)2017-03-06
