
中空碳纳米棒负载Fe3O4的制备及催化性能研究
2019-05-28 02:41孟庆男崔娟妮
西安理工大学学报 2019年1期
孟庆男,崔娟妮,赵 康
(西安理工大学,材料科学与工程学院,陕西西安710048)
近年来,随着经济的不断发展,我国的水污染问题日益严重,对国民健康和社会发展构成了严重威胁。Fenton技术是利用H2O2分解产生的 ·OH来降解水中有机污染物的一种方法,由于其绿色、高效,受到了研究人员的广泛重视[1]。Fe3O4纳米材料作为一种常用的非均相Fenton反应催化剂,不仅活性高,还克服了均相Fenton反应中易产生铁污泥、铁盐无法重复利用等缺点。但磁性纳米粒子存在不稳定、易团聚、容易被氧化等缺点,限制了其催化性能的发挥[2]。目前,常用的方法是将Fe3O4负载在惰性载体中。对于载体,不但要起到防止Fe3O4纳米材料聚集的作用,还需保证水中有机污染物能到达催化剂表面[3-6]。
碳在常温下非常稳定,能够耐强酸、强碱,与有机物及氧化物相比更适合作为纳米催化剂的载体[3]。此外,中空结构碳材料具有比表面积大、密度小,以及良好的吸附性和通过性,已被广泛应用于催化领域[7]。因此,本文选取商用Fe2O3纳米棒为前驱体,利用“一锅法”实现二氧化硅和间苯二酚-甲醛树脂的包覆,随后通过热处理同时实现树脂壳层的碳化和内部Fe2O3的还原,最终制备中空碳纳米棒负载Fe3O4材料(Fe3O4@h-C)。所制备的Fe3O4@h-C在亚甲基蓝的催化降解反应中显示出良好的催化活性和重复使用性能。中空结构不但有利于阻止纳米Fe3O4的聚集,还能保证其与有机污染物充分接触,有利于Fe3O4材料催化性能的提升。
1 实验
1.1 试剂与仪器
Fe2O3纳米棒,购自阿拉丁试剂公司。无水乙醇、氨水(质量分数25%)、正硅酸乙酯(TEOS)、间苯二酚、乙醇、甲醛、NaOH均为分析纯,购自国药集团化学试剂有限公司。实验用水为去离子水。
JEOL-2010型透射电子显微镜(TEM),日本电子株式会社;Nicolet Avatar 360型红外光谱仪(FTIR),PerkinElmer公司;Lambda 750型紫外-可见分光光度计(UV-Vis),PerkinElmer 公司;SiemensD-5005型X射线衍射仪(XRD),Bruker公司;DTG-60H型热重分析仪(TGA),日本岛津公司。
1.2 催化材料的制备
称取0.479 g Fe2O3纳米棒,加入到32 mL去离子水、128 mL乙醇和3 mL氨水的混合液中,搅拌30 min后加入1 mL正硅酸乙酯(TEOS)。10 min后,向溶液中加入0.2 g间苯二酚,再隔10 min加入0.28 mL甲醛。磁力搅拌8 h后,将产物离心、洗涤、干燥,所得产物在5 %氢气、95 %氮气的气氛中500 ℃还原、碳化,保温2 h。最后用1 mol/L的NaOH刻蚀SiO2中间层,得到Fe3O4@h-C纳米棒。作为对比,在制备过程中不加TEOS制备四氧化三铁-碳核壳型(Fe3O4@C)纳米棒;在制备过程中不加间苯二酚和甲醛制备纯Fe3O4纳米棒。
文章中关于粒径和尺寸的数据,均通过Photoshop软件测量TEM照片得到,每次测量不少于20组数据,然后取平均值。
1.3 催化性能测定
将200 mL(50 mg/L)亚甲基蓝溶液调节到指定温度和pH 值,然后称取20 mg的催化剂样品分散于溶液中,搅拌30 min至吸附平衡,加入一定量的双氧水溶液(30%);通过紫外-可见分光光度计对亚甲基蓝溶液在665 nm处的吸光度进行检测,并计算其降解率[1]。
2 结果与讨论
2.1 催化材料的结构与形貌
图1是在Fe2O3纳米棒表面包覆二氧化硅和间苯二酚-甲醛树脂后,产物在氮气下的TGA测试结果。从曲线上可以看出,失重是分两个阶段的。第一阶段是200 ℃以下,这段的失重主要是因为样品中的少量水分和其他挥发性的小分子杂质的损失;第二阶段是从200 ℃开始的,在这个阶段,树脂逐渐碳化,当温度上升到800 ℃以上时,曲线趋于直线,此时样品的失重率约为17%[8]。根据TGA曲线,样品的热处理温度选择500 ℃,既可保证树脂的碳化,又能防止Fe3O4在高温下转化为单质铁[9]。

图1 包覆产物的TGA曲线Fig.1 TGA curve of the coated Fe2O3 precursor
图2(a)为所采用的商用Fe2O3纳米棒的XRD图谱,其衍射峰与斜方六面体晶型α-Fe2O3的标准卡片(JCPDS No.87-1164)相符,没有杂质相,说明其纯度较高[10]。经过热处理及NaOH刻蚀后,最终产物的XRD谱图(图2(b))中,在2θ为 30.1°、35.7°、43.3°、57.2°、62.8°处的衍射峰分别对应面心立方Fe3O4晶体(JCPDS No.75-0033)的(220)、(311)、(400)、(511)、(440)晶面,这表明Fe2O3已完全转化为Fe3O4,且在刻蚀的过程中没有发生明显的氧化[11]。此外,谱图中没有出现石墨的衍射峰,说明碳化温度较低,碳以无定型形式存在[9]。

图2 Fe2O3和Fe3O4@h-C的XRD谱图Fig.2 XRD patterns of Fe2O3 and Fe3O4@h-C
图3(a)显示,Fe3O4@h-C的氮气吸附脱附等温曲线是典型的第IV类等温线,表明产物中存在大量介孔,在P/P0(相对压力)为0.5~0.9范围内,曲线上存在一个明显的滞后环,这是由于其空腔结构对氮气的吸附造成的[12]。从图3(b)可以看出,产物的孔尺寸主要在10 nm左右。此外,Fe3O4@h-C的BET比表面积高达119.1m/g,有利于提高其催化活性。
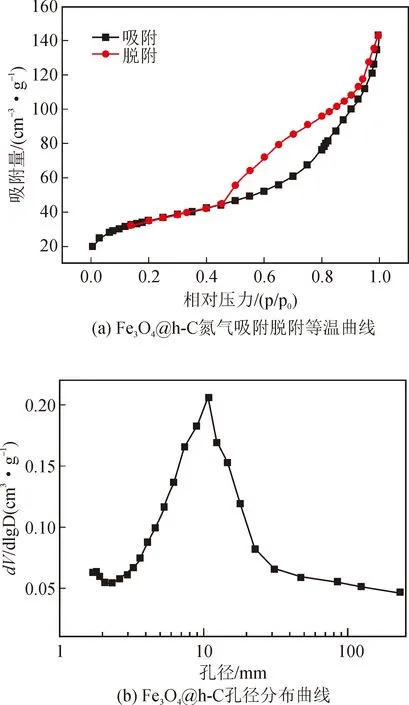
图3 Fe3O4@h-C的氮气吸附脱附等温曲线和孔径分布曲线Fig.3 Nitrogen adsorption desorption isotherm curve and pore size distribution curve of Fe3O4@h-C
从TEM照片中(图4(a))可以看出,作为原料的Fe2O3纳米棒,其平均长度约为63.0 nm, 宽度为18.7 nm。将该Fe2O3纳米棒进行二氧化硅、间苯二酚-甲醛树脂包覆并热处理之后,从产物(Fe3O4@SiO2@C)的TEM照片(图4(b))中可以观察到样品具有明显的核壳结构。其中,Fe3O4纳米棒全部位于产物的中间位置,其尺寸约为长43.6 nm,宽14.2 nm,略小于图4(a)中的Fe2O3,说明在还原过程中发生了体积收缩[13]。此外,产物的壳层厚度约为12.8 nm,但由于碳和二氧化硅具有相似的衬度,二者之间观察不到明显的界面[1]。如图4(c)和(d)所示,经过NaOH刻蚀后,产物都转换成 “铃铛型”结构,说明成功制备了Fe3O4@h-C。Fe3O4@h-C外部的碳壳层厚度约为3.7 nm,并没有发生变形和塌陷,在有效保护内部Fe3O4的同时,还有利于内外物质的快速通过[1]。值得注意的是,产物内部的Fe3O4偏离了中心位置,说明其在壳层内部具有一定活动能力,有利于催化反应中与有机物充分接触[14]。

图4 Fe2O3、Fe3O4@SiO2@C和 Fe3O4@h-C的TEM图像Fig.4 TEM images of Fe2O3, Fe3O4@SiO2@C and Fe3O4@h-C
图5是刻蚀前后,样品(Fe3O4@SiO2@C与Fe3O4@h-C)的红外特征谱图。两种样品在3000~3700 cm-1处是-OH基团的伸缩振动吸收峰,在1600 cm-1左右的吸收峰来自于苯环骨架的振动,在1400 cm-1左右的吸收峰为-CH2-剪切振动吸收峰,在580 cm-1处的吸收峰来自于Fe-O键的伸缩振动[4, 8]。对于刻蚀前的Fe3O4@SiO2@C,其谱图中位于1095 cm-1处的吸收峰来自于Si-O-Si的反对称振动[1];对于刻蚀后的Fe3O4@h-C纳米棒,其谱图中归属有机基团和Fe3O4的峰依然可见,而归属二氧化硅的两个峰基本消失了,且在1056 cm-1处出现了新的来自于C-O键的伸缩振动吸收峰[8]。以上结果表明,刻蚀过后,二氧化硅已被除去,碳化过后的样品中还有大量的有机基团,这有利于其在水中的分散,有利于催化反应的进行。
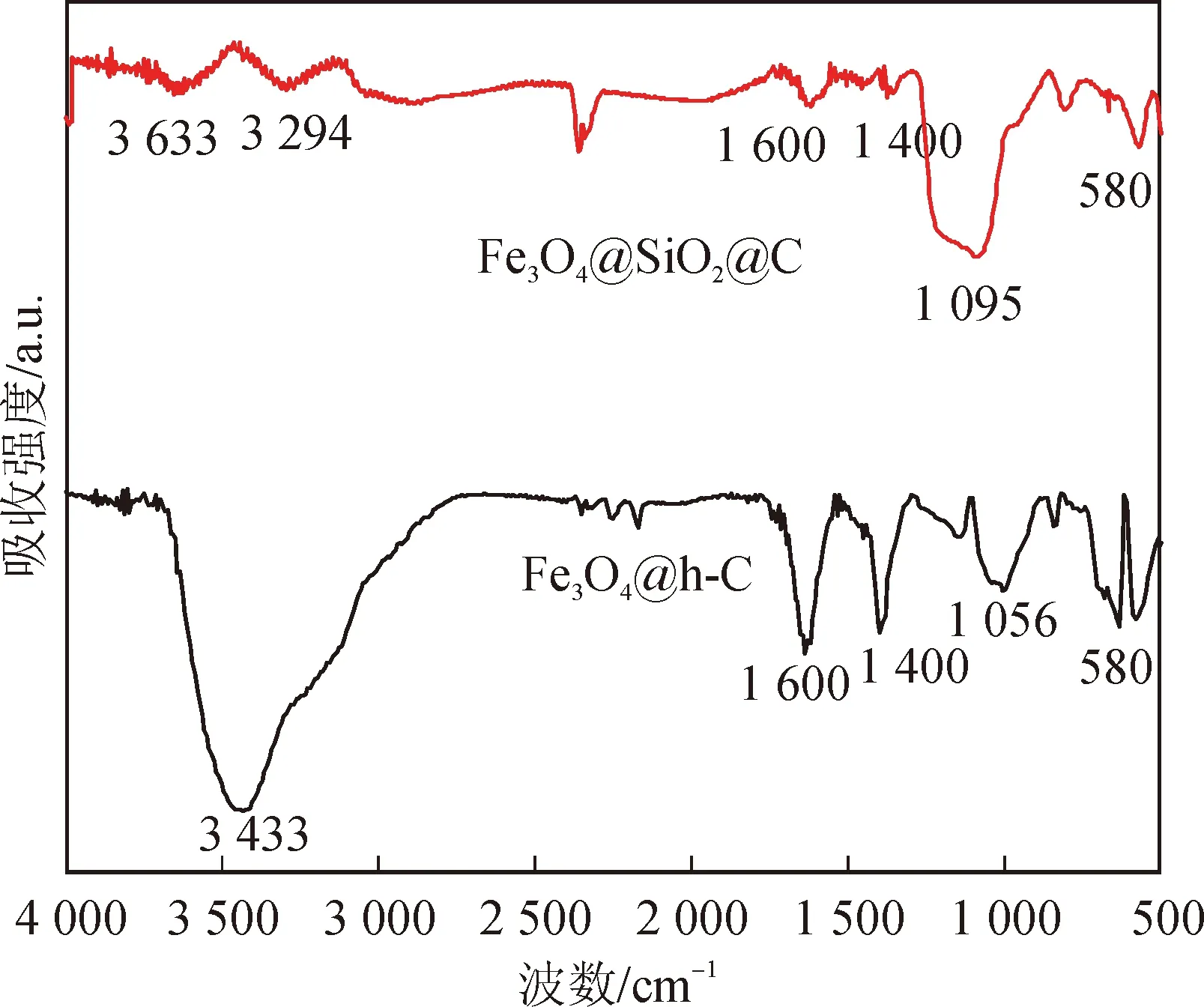
图5 Fe3O4@SiO2@C和Fe3O4@h-C的傅里叶红外谱图Fig.5 FTIR of Fe3O4@SiO2@C and Fe3O4@h-C
为了考察材料结构对催化性能的影响,本研究还分别制备了Fe3O4@C纳米棒和纯Fe3O4纳米棒(图6(a)和(b))。从相应的XRD数据(图6(c))可以看出,两个试样中都存在着结晶性良好且不含杂质的面心立方Fe3O4晶体,这与Fe3O4@h-C纳米棒的结果相一致。值得注意的是,从TEM照片可以看出,Fe3O4@C纳米棒的碳壳层厚度约为15.7 nm,要大于Fe3O4@h-C纳米棒的碳壳层厚度。
2.2 磁性测定
图7(a)给出了室温下Fe3O4@h-C、Fe3O4@C及Fe3O4三种材料的磁滞回线。从图中可以看出,三种材料的磁滞回线均不通过原点,其矫顽力分别为155、242、129 Oe,呈弱铁磁性[3]。由文献报道可知,当Fe3O4的尺寸超过30 nm后,其磁性将由超顺磁性向铁磁性转变,这与TEM照片的结果是相符的[15]。Fe3O4@h-C、Fe3O4@C及Fe3O4纳米棒的比饱和磁化强度(Ms)分别为58.5、65、45.7 emu/g。值得注意的是,由X射线能谱分析(EDS)可知,Fe3O4@h-C和Fe3O4@C中的Fe3O4含量分别为62.6 % 和 49.1 %,但是其Ms均高于纯的Fe3O4。这可能是由于聚合物分解会形成独特的微环境,使产物的磁性增强。不过由文献可知,以上三种材料的Ms均较高,可以保证样品具有较强的磁响应能力[2-4]。以Fe3O4@h-C为例(图7(b)),在一块儿钕铁硼永磁体的作用下,数秒钟即可实现从溶液中的分离。

图6 Fe3O4@C、Fe3O4的TEM图像和 Fe3O4@C、Fe3O4的XRD谱图Fig.6 TEM images of Fe3O4@C, Fe3O4 and XRD patterns of Fe3O4@C, Fe3O4

图7 Fe3O4@h-C的磁滞回线和磁分离效果照片Fig.7 Hysteresis loop and magnetic recycling performance of Fe3O4@h-C
2.3 催化性能测定
由图8(a)可知,在反应温度T=50 ℃, pH = 3,搅拌30 min至吸附平衡,加入0.8 mmol/L H2O2的反应条件下,以Fe3O4@h-C为催化剂,加入H2O2仅5 min,亚甲基蓝(MB)的褪色率Ct/C0(C0为亚甲基蓝溶液初始浓度,Ct为t时刻亚甲基蓝溶液浓度)就可以达到95 %;10 min后, MB的褪色率就可达到97.9 %。在同等条件下,纯Fe3O4纳米棒对于亚甲基蓝的褪色率分别为79 %(5 min)和91 %(10 min),低于Fe3O4@h-C。而对于Fe3O4@C, 即使将反应时间延长到30 min,其对MB的降解也仅有21 %。这主要是由于Fe3O4@h-C具有独特的中空结构,能够保证溶液中的亚甲基蓝分子与内部的Fe3O4颗粒充分接触,并使得催化剂附近亚甲基蓝浓度增加,有利于加快反应速率[1]。相反,对于Fe3O4@C纳米棒,较厚的碳壳层阻碍了Fe3O4与溶液中亚甲基蓝分子的接触。而纯Fe3O4纳米棒在溶液中容易聚集,因此催化性能也低于Fe3O4@h-C。此外,在没有催化剂的情况下,H2O2自身对MB的褪色率较低;而没有H2O2加入的情况下,Fe3O4@h-C纳米棒对MB基本没有褪色作用。由此可见,反应是按照Fenton反应机理进行的[8]。
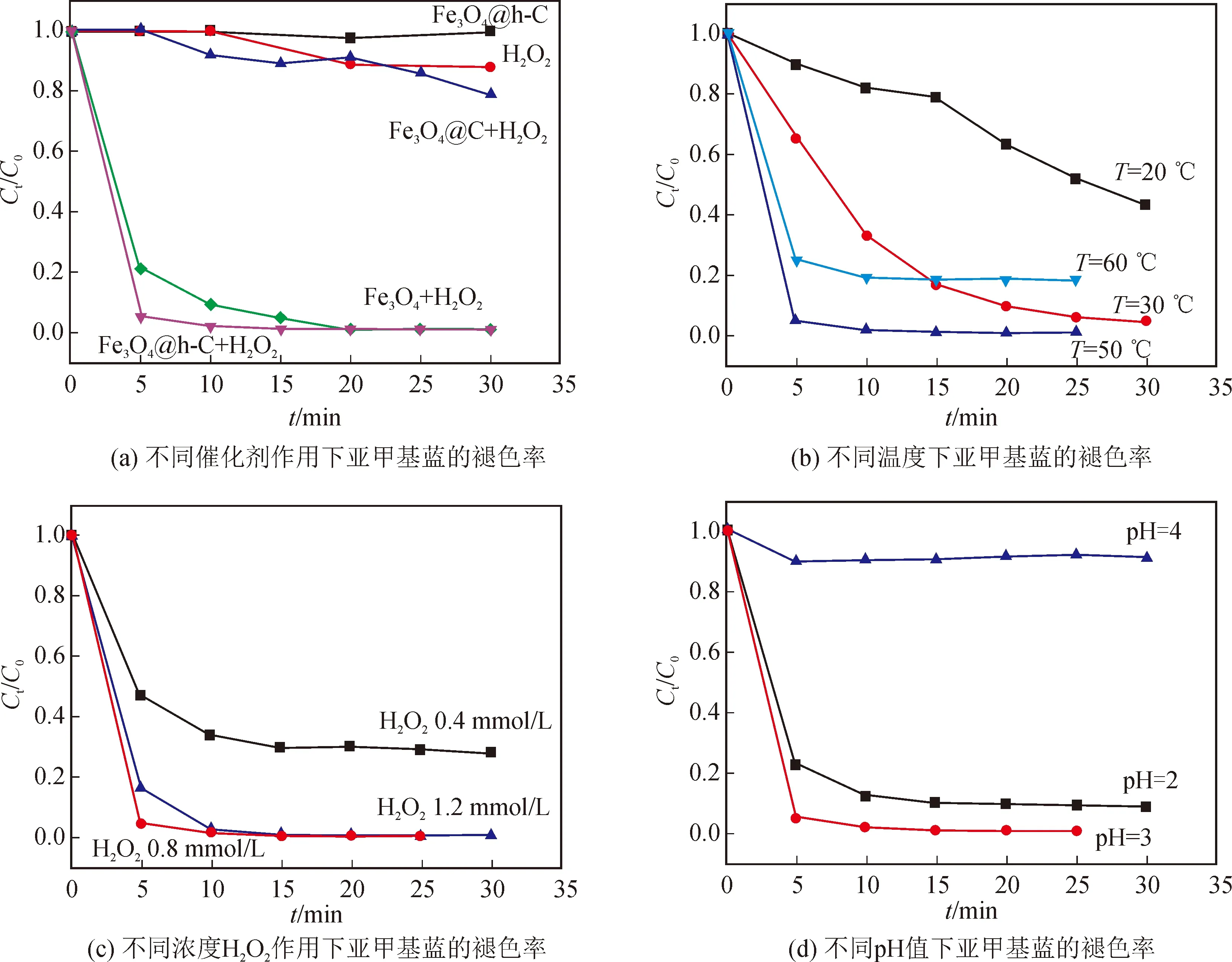
图8 不同条件下Fe3O4@h-C对亚甲基蓝的褪色率Fig.8 Fading rates of MB of Fe3O4@h-C under different conditions
接下来,在保持其它反应条件不变的情况下,分别调节温度、双氧水浓度和pH值,探讨其对亚甲基蓝褪色率的影响。图8(b)是不同温度条件下Fe3O4@h-C对亚甲基蓝的褪色率,在低于50°C时,MB的褪色速度随着温度的升高而加速,而当温度升高到60°C时,褪色率反而下降,这有可能是由于过高的温度导致H2O2加速分解,从而降低了反应速率。图8(c)是不同浓度H2O2条件下,Fe3O4@h-C对亚甲基蓝的褪色率,当H2O2浓度较低(0.4和0.8 mmol/L)时,亚甲基蓝的降解率随H2O2浓度的增加而增加;当H2O2浓度大于0.8 mmol/L时,降解率反而有所下降。这是因为在H2O2浓度较低时,H2O2浓度的增加会产生更多的羟基自由基,促进类Fenton反应;而在高H2O2浓度下,过量的H2O2则充当羟基清除剂将羟基自由基转化为活性较低的自由基,导致降解率下降[12]。图8(d)是不同pH值条件下,Fe3O4@h-C对亚甲基蓝的褪色率,可以发现,反应的最佳pH值为3。当pH值增加到4及以上时,亚甲基蓝的降解率急剧下降,这有可能是因为生成了无活性的离子(FeO2+);当pH值降低为2时,亚铁离子会形成活性较低的铁络合物,并且高浓度的H+与H2O2形成稳定的[H3O2]+,减少了羟基自由基的产生,从而降低了亚甲基蓝的降解率[12]。图9是Fe3O4@h-C纳米棒的循环催化实验结果。由图可知,Fe3O4@h-C纳米棒表现出良好的稳定性,在循环使用四次后,MB的降解率依然可以达到92.8 %,表现出了良好的重复使用性,易于回收再利用。

图9 Fe3O4@h-C的循环催化反应结果Fig.9 Recycling results of the Fe3O4@h-C nanorod catalyst
3 结 论
本文以商用Fe2O3纳米棒为起始原料,通过溶胶凝胶法和氢气还原的方法制备了Fe3O4@h-C纳米棒。产物具备磁分离能力。在催化H2O2降解亚甲基蓝的反应中,Fe3O4@h-C纳米棒展现出良好的催化性能和重复使用性,显示出一定的实际应用价值。通过对比发现,中空结构不但有利于阻止Fe3O4的聚集,还能保证其与有机污染物充分接触,对Fe3O4纳米棒催化性能的提高是十分有益的。
猜你喜欢
青少年科技博览(中学版)(2022年11期)2023-01-07
分子催化(2022年1期)2022-11-02
化学工业与工程(2022年1期)2022-03-29
少儿科技(2022年2期)2022-03-05
粉末冶金技术(2021年3期)2021-07-28
中成药(2019年12期)2020-01-04
天然产物研究与开发(2019年10期)2019-11-05
中国塑料(2016年2期)2016-06-15
中国民族医药杂志(2016年2期)2016-05-14
云南师范大学学报(自然科学版)(2015年5期)2015-12-26
