
GPA:A Microbial Genetic Polymorphisms Assignments Tool in Metagenomic Analysis by Bayesian Estimation
2019-05-14 10:07:32JiaruiLiPengchengDuAdamYongxinYeYuanyuanZhangChuanSongHuiZengChenChen
Jiarui Li,Pengcheng Du ,Adam Yongxin Ye,Yuanyuan Zhang Chuan Song ,Hui Zeng *,Chen Chen *
1 Beijing Key Laboratory of Emerging Infectious Diseases,Institute of Infectious Diseases,Beijing Ditan Hospital,Capital Medical University,Beijing 100015,China
2 Center for Bioinformatics,State Key Laboratory of Protein and Plant Gene Research,School of Life Sciences,Peking University,Beijing 100871,China
KEYWORDS Next-generation sequencing;Pool-seq;Bayesian model;Metagenomics;Genetic polymorphisms
Abstract Identifying antimicrobial resistant(AMR)bacteria in metagenomics samples is essential for public health and food safety.Next-generation sequencing(NGS)technology has provided a powerful tool in identifying the genetic variation and constructing the correlations between genotype and phenotype in humans and other species.However,for complex bacterial samples,there lacks a powerful bioinformatic tool to identify genetic polymorphisms or copy number variations(CNVs)for given genes.Here we provide a Bayesian framework for genotype estimation for mixtures of multiple bacteria,named as Genetic Polymorphisms Assignments(GPA).Simulation results showed that GPA has reduced the false discovery rate(FDR)and mean absolute error(MAE)in CNV and single nucleotide variant(SNV)identif ication.This framework was validated by whole-genome sequencing and Pool-seq data from Klebsiella pneumoniae with multiple bacteria mixture models,and showed the high accuracy in the allele fraction detections of CNVs and SNVs in AMR genesbetween two populations.Thequantitativestudy on thechangesof AMR genesfraction between two samples showed a good consistency with the AMR pattern observed in the individual strains.Also,the framework together with the genome annotation and population comparison tools has been integrated into an application,which could provide a complete solution for AMR gene identif ication and quantif ication in unculturable clinical samples.The GPA package is available at https://github.com/IID-DTH/GPA-package.
Introduction
Bacterial antimicrobial resistance is considered as‘‘one of the biggest threats to global health,food security,and economic development today”by the World Health Organization(WHO).The effective prevention and treatment of infections caused by bacteria require increasingly greater monitoring and prevention activities[1].Traditional bacterial monitoring methods by the culture technology followed with the classif ication by genotyping such as multi-locus sequence typing(MLST)[2]and multi-locus VNTR analysis(MLVA)[3],and pulsed-f ield gel electrophoresis(PFGE)[4],have been used for morethan thirty yearsto determinecausativeagentsin outbreaks and track the epidemiological trends[5].However,at present,the culture technology and large genomic diversity within thespecies limited thedevelopment of these monitoring methods.For example,only 126 bacteria have available typing schema for MLST test[6-8].This limitation resulted in a 20%shortfall of global outbreaks that could not be adequately illustrated[9].Improving the genotyping method is an urgent need for better monitoring and epidemiological surveillance.
Next-generation sequencing(NGS)technology has provided immense genotyping data for observing rare and lowfrequency genetic variations in complex samples for precision medicine[10].In humans and other mammals,these data are well employed to observe molecular variations such as copy number variation(CNV)and single nucleotide variant(SNV)[11].A seriesof bioinformatic tools have been developed,such as BreakSeek[12],STRiP[13],SVM2[14],LUMPY[15],and inGAP-sv[16].Among them,STRiP is well known for CNV detection[13],while GATK[17]and SAMtools[18]tools are used for SNV identif ication.However,the model f itting process of these tools was based on a diploid genome,such as human or mouse genome.Unlike mammals,the clinical samples are complex samples with a large amount of bacteria presenting a large genomic diversity,even within the same species[19].These tools thus showed a large bias in quantifying the proportion of genes from single species.Also,some metagenomics analysis tools have also been developed to investigate the molecular variations in complex samples,to measure bacterial diversity and abundance,or to identify functional changes in microbial communities under a species level[20].However,given the lack of an eff icient algorithmic model to quantify the identif ied genes in complex samples,we have few bioinformatics tools to continuously observe the dynamic and potential quantitative changes in a given bacterial population.
The Bayesian model is a widely used algorithm to estimate bacterial diversity and abundances in complex NGSdata samples for its strong capability to estimate genotypes in spite of sequencing errors[21,22].These errors may be generated for several reasons[13]:molecular libraries contain chimeric molecules that may be misidentif ied as structural variants[23];read depths vary across the genome in ways that they also vary among different sequencing libraries[24];and alignment algorithms are misled by the tandem repeats in the genome[25].GATK,developed by Broad Institute,has been applied to reduce the impact of the errors in human and mouse sequencing data.For complex bacterial samples,this software may be used with a polyploidy algorithm to estimate SNV proportions[17].However,GATK has rarely been evaluated in these samples.In addition,a Bayesian model pipeline for estimation of CNVs in bacteria is still lacking,with the exception of Breseq that can only be used for one genome at a time[26].
Here,we have developed the Genetic Polymorphisms Assignments(GPA),a Bayesian framework for genotyping multiple bacterial in mixed samples,including a genome annotation pipeline.To evaluate the accuracy of GPA,we compared Pool-seq data and individual genomic data of Klebsiella pneumonia,and found that GPA could(i)detect all SNVs and CNVs in Pool-seq data,and(ii)calculate an accurate frequency of each known allele of target genes,which were identif ied by individual genomic data.We further demonstrated its capabilities through a consecutive analysis on the frequency of the tolC gene,as well as rmpA and rmpA2,which we previously reported in CR-HvKP,showing sequence analysis consistent with phenotypic changes over two years.This software package can also be used to monitor antimicrobial resistant genes in metagenomics data for a specif ic pathogen.
Results
Performance evaluation by simulation studies
Overview of the GPA package
The workf low of the GPA package is shown in Figure 1.First,we processed the BAM f ile with the standard GATK pipeline:raw reads were mapped with BWA-mem and duplicates were removed with Picard(Figure 1A).Then,the BAM f iles were processed in the novel CNV calling pipeline model.Using a Bayesian framework,GPA analyzes the coverage depth at each position and predicts its ploidy type(Figure 1B).The results of this ploidy assignment were used as input for the further identif ication of SNV with GATK Unif iedGenotyper calling(Figure 1C).The total CNV and SNV results were then mapped to reference genes and annotated for functional prediction and population analysis(Figure 1D).This pipeline serves as a complete toolkit for genotype assignments in pooled bacterial sequence data.The GPA package is available at https://github.com/IID-DTH/GPA-package.
GPA model applied to a simple CNV example
To better illustratethe GPA model when used in a CNV study,we used a mixed dataset consisting of three randomly selected Klebsiella pneumoniae genomes(Kpn12,Kpn14,and Kpn32)as Pool-seq data to evaluate the model,posterior of different genotypes,and major alleleassignment(Figure2A-E).Considering thepreviousbioinformatics tools preferred to applying in mammals with a diploid sample,we used a mixture with three samples as a simple model,which also tends to be a common occurrence in metagenomes.We used this model to show deletion,normal,or insertion states with a 0,1,or 2 in each reference genome position(Figure 2A).Seven allele types were thus presented for each position in Pool-seq data,including 0,1/3,2/3,1,4/3,5/3,and 2.(Figure 2A).

Figure 1 Schematic overview of the GPA packageA.The process of data mapping.B.The CNV calling model:the depth in each position is analyzed(upper panel)and predicted for its ploidy number with a Bayesian model(down panel).The pink and blue lines in the circle indicate duplication and deletion regions,respectively.C.The SNV calling and correction model:theploidy analysisresult and processed BAM f ileareused asinput for SNV calling and identif ication with GATK Unif ied Genotyper.The allele fraction is corrected using the CNV identif ication result.The fractions of the alternateand referencealleles areindicated in red and yellow,respectively.Thegray bar indicate deletion regions.D.The CNV and SNV calling results are annotated using the reference genes for functional prediction and further population analysis.The orange arrow representsa transcript in the genome.The mutants in thepopulation aredrawn with yellow or pink circles.CNV,copy number variation;SNV,single nucleotide variation.
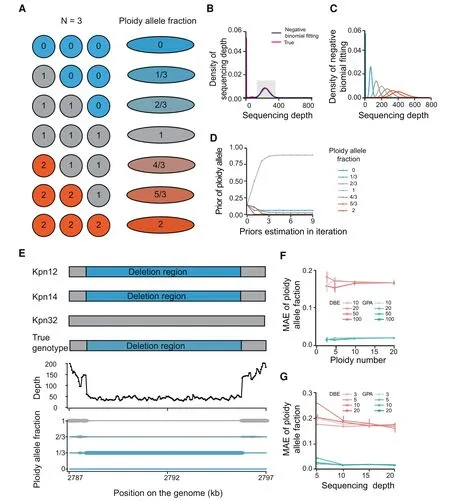
Figure 2 CNV model in the GPA packageA.Diagram of how ploidy allelefraction iscalculated using an exampleploidy(n=3).Blueindicatesdeletion,and red indicatesduplication.‘‘1′′isa normal stagewith amixtureof 3 bacteria.B.Thenegativebinomial distribution f itting themain distribution curveof coveragedepth for each nucleotidesite.Thered linerepresentsthereal depth distribution in a simulation using the Pool-seq data from 3 genomes.Theblack linerepresentsthef itted negativebinomialdistribution,and theshadow region representsthemain peak.C.Read depth modeled by anegative binomial distribution with different allelefractions.Seepanel A for color codes.D.Prior estimation using iterations.E.An exampleof GPA used to identify Kpn deletion regionsin Pool-seq data.Theindividual genomeshotgun sequencingdatafor Kpn12and Kpn14haveadeletion in thegenomeindicated by bluerectangles.Thesizeof thisdeletion in thebottom panel representstheestimated posterior for different allele types.F.MAE calculated with different ploidy numbersusing GPA(green)and traditional DBA(red).Thecolor gradient of thelines(from light to dark)representsthecoveragedepth in themodel of 10×,20×,50×,and 100×,respectively.G.Evaluation of GPA performancein low sequencingdepth.MAEwascalculated with differentcoveragedepthsusing GPA(green)and traditional DBA(red).Thecolor gradient of the lines(from light to dark)representsthenumber of ploidiesin themodel,which are3,5,10,and 20,respectively.MAE,mean absoluteerror.
The Pool-seq data were aligned to the reference genome,and f itted thereadsdepth with a negativebinomial distribution curve and estimated its parameterκ(Figure 2B).Using this parameter,we calculated the likelihood value of each modeled negativebinomial distribution in each possiblealleletype(Figure 2C).Next,the posterior were estimated by multiple iterations,and we observed that the priors converged quickly in 3 iterations(Figure 2D).The major allele assignment was displayed with 3 individual genome structures and compared with mixed data.We found that both Kpn12 and Kpn14 genomes had a~170-bp deletion,while Kpn32 was in normal stage.This result was consistent with the observation in mixed data,which wasassigned with thepeak valueof alleletypeaccording to the posterior(Figure 2E).
Performance evaluation of CNV analysis by simulation studies
Weexpanded the mixture analysis from 3 genomesto multiple genomes(>3).We used genomic data from 2009 and 2013 from 44 K.pneumoniae isolates as a control,with shotgun reads having an average depth of 150×as a simulated data pool.24 sample data were generated by constant coverage(5,10,15,20,50,and 100×,respectively)from 3,5,10,and 20 randomly selected samples.To improve robustness,we repeated to generate three replications for each sample data.The mean absolute error(MAE)of the control was used to measure the performance of different estimation methods.
We compared the performance of GPA with the previous depth-based estimation(DBE)approach.Both approaches readily identif ied deletions and duplications that occurred in most strains,however,more errors were introduced when deletions or duplications occurred in only a few strains(i.e.,the allele frequency was near to the peak)(Figure S1).In this condition,the GPA method had fewer false positive results in the detection of deletions or duplications.The average MAE was 0.02 for GPA and 0.2 for DBE.To evaluatetheeffect of depth,we simulated an average depth of 10,20,50,and 100×.The results showed that the GPA method was more tolerant of low depth datasets(Figure 2F).For depths of more than 20×,the detection was relatively stable.To evaluate the effect of ploidy number in low depth datasets,we simulated ploidies of 3,5,10,and 20 under the depth of 20×.The MAE was relatively dispersed when the ploidy number was small(Figure 2G).The GPA method was more robust compared to the DBE method in both low depth and small ploidy number.In total,the GPA method detected polymorphisms more accurately.
GPA model with a simple SNV example
To better illustrate the GPA model when used in an SNV study,we presented an example of SNV with 10 ploidies(Figure 3).Notably,in this model,f ive genomes had a deletion in the middle of the sequence.Traditional SNV calling tools always ignore to correct the mutation frequencies,when the mixed samples have deletions or duplications in the given genome site.In this example,there were f ive deletion alleles in a particular position,two reference alleles and three alternative alleles;the traditional SNV caller reported four reference alleles and six alternative alleles,determining a ploidy level of 10 in this position.The detected alternative allele fraction(6/10)would be higher than the true state(3/10),leading to an incorrect conclusion.In contrast,the duplication would lead to a relatively lower estimate of the allele fraction.From the previous results of CNV identif ication in our simulation,the deletion region ranged from 10%to 15%of the genome size.Thus,a more accurate model for SNV identif ication is required.
Performance evaluation of SNV analysis by simulation studies
To evaluate the performance of SNV analysis of GPA,we use the same simulated data as in the CNV analysis,and f irstly assessed the relationship between MAE and deletion fraction in 10 ploidy states with 50×coverage.At the whole genome level,using the GATK and DBE methods,we observed a signif icant increase in MAE of SNVs when the deletion allele fraction increased.In the same analysis,the GPA method performed better in deletion identif ication,which corrected most of the CNVs when the deletion fraction was over 0.3,thus resulting in a sharp decrease in MAE(Figure 4A).We also measured the accuracy of the frequencies of alternative alleles and found the accuracy was also signif icantly increased(Figure 4B).Comparison of results for other states also showed signif icant improvements in accuracy when CNV was considered in the model(Figures S2 and S3).
We also measured the combined impact of sequencing depth and ploidy number on MAE in SNV analysis.The MAE value showed a steep decrease and kept a constant low level when the depth increased to 20(Figure 4C).Interestingly,with the increasein ploidy number,the GPA method presented a lower MAE(Figure 4D),which suggested that this method was more suitable for the identif ication of complex samples than the traditional GATK and DBE method.
Performance evaluation in complex samples
Identif ication of CNVsand SNVs from Pool-seq data and public metagenomics data
To evaluate the potential of GPA in the discriminant analysis in real datasets,we used two datasets,(1)Pool-seq data from 44 K.pneumoniae strains,which could be divided into two groups with the same number(one from 2009,and the other from 2013),and(2)public metagenomics data from threestudieson thehuman gut microbiota[27-29].In the GPA package,we integrated genome mapping,CNV and SNV identif ication,as well as the variant annotation system.
We evaluated the capability of GPA to discriminate between individual genomes within the Pool-seq data.Using this procedure,we detected 18,131 and 27,819 deletion regions in 2009 and 2013 datasets,respectively,from a total of 9.29 G genome data(~2000×).Compared to the reference genome,our pipeline observed 10.3%and 10.6%of genomic deletion regions in the data from 2009 and 2013,respectively.Due to the lack of comparable approaches to identify bacterial SNVs and CNVs in complex data,we compared these deletions to the deletion regions which we identif ied in the alignments of the individual genome to the reference genome data.These deletions covered 23.02%of deletion regions which were observed in the individual genome from 2009 and 60.05%of deletion regions observed in the individual genome from 2013.We also identif ied 1154 and 216 duplication regions in the dataset from 2009 and 2013,respectively(Figure 5A).Compared to thenumber of identif ied deletionsin thegenome,the number of duplications was limited.SNVs were also identif ied in these two separate datasets.We observed 692,368 and 272,169 SNV sites in the dataset from 2009 and 2013,respectively,which were similar to the SNV detection by GATK(698,528 and 275,174 sites,respectively).Further analysis of the SNV site coverage to actual genome data presented similar coverage of 79%in the dataset from 2009,but with different MAE rates.In datasets from 2009 and 2013,the MAE rates were 2.00 and 1.78,respectively,when using GATK,which were 1.43 and 1.40,respectively,when using GPA,indicating a lower error rate with the GPA package.

Figure 3 SNV correction model in the GPA packageGenomic featureof 10 sequencing readsin a given genomic site.Theyellow circlesrepresent siteson thisgenomethat havethesameallele as thereferencegenome‘‘T”,the red circlesrepresent alternativealleles,for example‘‘A”,and theblue circlesrepresent deletion sites.Ref,reference allele;Alt,alternative allele;Del,deletion allele.
We further evaluated the ability of GPA to discern SNVs within the Pool-seq data by comparing results obtained using GPA and GATK.In the different genetic sites observed by individual genome analysis on the comparison of 2009 with 2013 data,GPA identif ied 128,803 SNVs.Among them,GATK failed to predict 6675 SNV sites.For example,all six SNV sites in KPHS_45500 were not properly detected by GATK,because there was a 203-bp deletion in 10 of the 22 genome data from 2009(Table 1).This result suggested that the use of GATK in metagenomics or Pool-seq data from bacteria still has limitations,although it is very effective for data from humans and other mammals.
Last,we expanded the application of the GPA package to metagenomics data in order to identify dynamic changes in gene frequencies within a given genome.The GPA package treated metagenomics data as multi-strain pooled samples.To obtain high enough coverage for the detected species,we evaluated our ability to identify SNVs and CNVs in the dominant species.Using MetaPhlAn2[30],we found that the dominant species in 15 samples was Prevotella copri,with particularly high proportions in the MH0001,MH0005,and MH0018 samples.Using the P.copri reference genome(ASM15793v1),we applied the reference-based GPA package to identify CNVs,SNVs,and indel mutationsin thethreesamples.We identif ied 608,1245,and 1290 SNV sites in the three samples compared with the P.copri reference.Moreover,we identif ied totally 1,192,437-bp deletion regions and 26,593-bp duplication regions on MH0001,1,106,144-bp deletion regions and 353,830-bp duplication regions in MH0005,as well as 1,068,700-bp deletion regions and 12,581-bp duplication regions on MH0018.Here,limited by the complexity of the computation,we optimized the number of mixed strains in the same species(N)to 20 in order to balance the precision and eff iciency,since the running time will exponentially increase as N increases in GATK.Although we cannot evaluateall thedeletions,duplications,and insertions,we compared the number of resistance genes identif ied by Resistance Gene Identif ier[31]to what was identif ied by BLAST in a previous report.In MH0001,MH0005,and MH0018,respectively,there were 40,45,and 44 previously identif ied resistance genes[27].Among them,5,5,and 4 genes were from P.copri,which matched to the reference genome ASM 15793v1.In our GPA package,we identif ied 1,2,and 3 overlapping genes,respectively.We identif ied 15 AMR genes in reference genome ASM 15793v1.In addition,we also identif ied another 10,9,and 9 antibiotic resistance genes in these metagenomics data(Table S1).Moreover,we identif ied the deletion and duplication regions,and calculated the specif ic fraction of modif ied alleles in these regions.Among these AMR genes,4 genes,EF-Tu,rpoB,gyrA,and ileS,performed antibiotic resistance through mutations,none of which were identif ied in previous BLAST pipelines.
The application of the GPA package to population comparisons To better apply the GPA package in the comparison of two groups,we constructed a complete pipeline for genome annotation using reference genome data.In a comparison of two Pool-seq data,K.pneumoniae strains from 2009 and 2013,we screened the whole genome and found signif icantly different genomic regions,including 184,654-bp CNV regions and 128,803 SNVs.Among these CNVs and SNVs,79.7%and 90.9%,respectively,were from coding regions,which was similar to the percentage of coding regions in K.pneumoniae.Then,using an automated model of ANNOVAR[32]with the reference genome,we annotated SNVs with 8919 non-synonymous and 51,053 synonymous sites.Thus,we can quickly and easily learn the biological meaning of these differences since this is an unusual pipeline in its ease of application for the identif ication of gene function within large,complex datasets.

Figure 4 Evaluation of the GPA package and comparison with other approachesA.The correlation between MAE and deletion fraction in 10 ploidy states with 50×coverage using different methods.MAE rate was calculated under different deletion conditions.Red representsthetraditional DBE method,green representsthe GATK method,and blue represents the GPA method.B.The correlation between accuracy and deletion fraction in 10 ploidy states with 50×coverage using different methods.C.Thecorrelation between MAE and sequencing depths.The color gradient of thelines(from light to dark)represents ploidy number of 3,5,10,and 20 in themodel.D.Thecorrelation between MAE and different ploidy numbers.Thecolor gradient of the lines(from light to dark)represents coverage depth in the model spanning 10×,20×,50×,and 100×.
This package also permitted automated annotation of SNV sites and CNV regions identif ied by our pipeline in metagenomics data with the P. copri reference genome(ASM 15793v1).In the MH0001 dataset,608 SNV sites were identif ied;1245 SNV sites were found in MH0005,and 1290 SNV sites were found in MH0018.Of these mutations,43 sites in MH0001,78 sites in MH0005,and 64 sites in MH0018 were identif ied as functional SNV sites,including nonsynonymous mutations and stop-gain mutations.The mutations were located in 5 genes across the three samples.The CNV regions were annotated to 7 genes in MH0001,10 genes in MH0005,and 7 genes in MH0018.
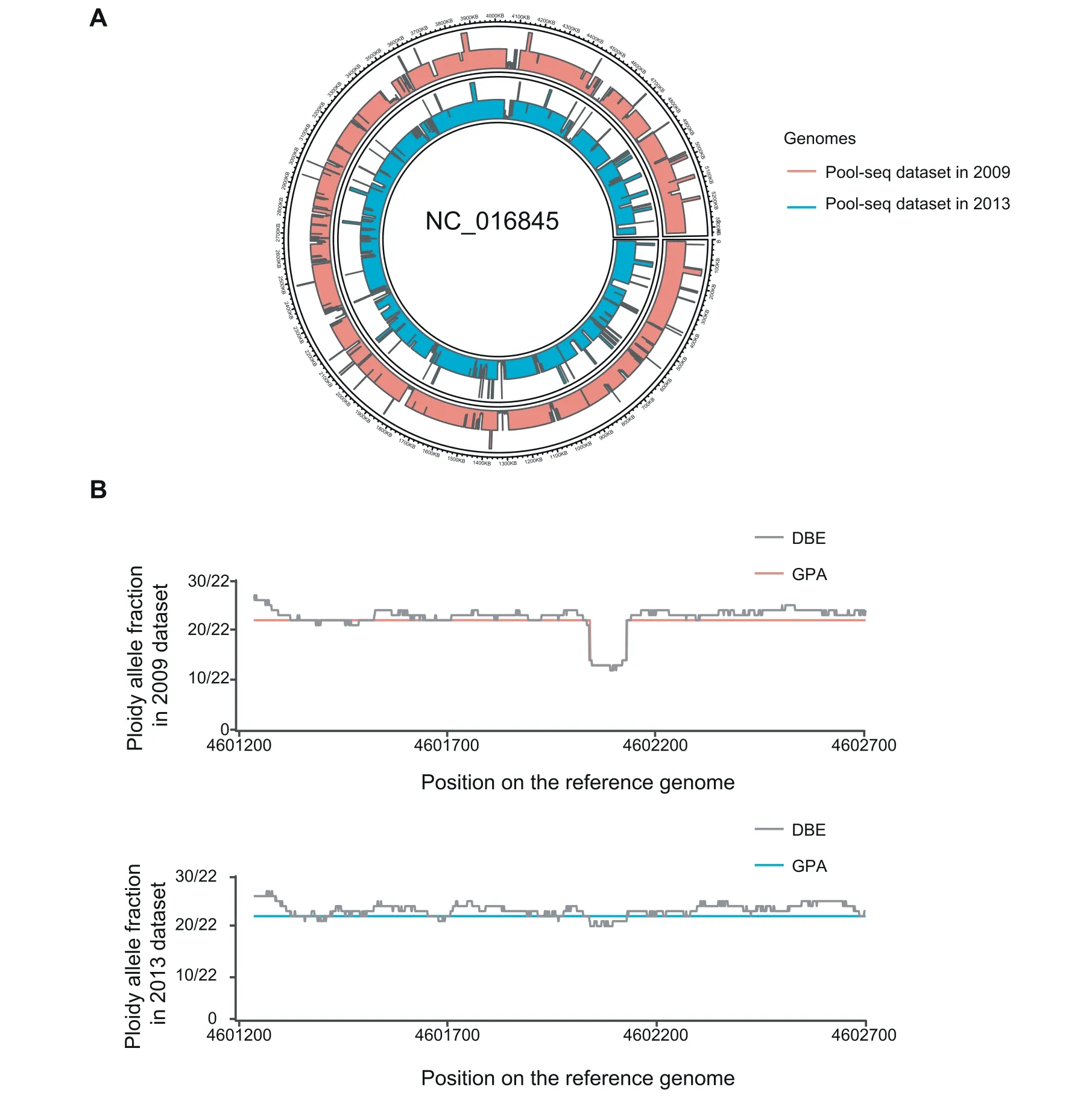
Figure 5 Comparison of Pool-seq data analyzed by GPA with individually analyzed data from 2009 and 2013 datasetsA.Identif ication of CNVsin Pool-seq data of K.pneumoniae in 2009 and 2013 datasetsby GPA.Thered block represents CNVsfound in the 2009 data and the cyan block represents CNVs in the 2013 data.B.Detection of deletions in the antimicrobial resistance gene tolC in K.pneumoniae populationsin 2009 and 2013 datasets.Thegray linerepresentstheresultsof DBE analysis;thered and cyan linesrepresent the results of GPA in 2009 and 2013 populations,respectively.
Validating the impact of mutations on virulence and antimicrobial resistance genes
The GPA model for the estimation of gene abundance was applied to Pool-seq datasets from 2009 and 2013 to evaluate thedynamic changes in thefrequencies on the functional gene.We compared the frequency of the virulent genes(rmpA and rmpA2)and antibiotic resistant gene(tolC)to two Pool-seq datasets.Among them,rmpA and rmpA2 are genes recently validated in CR-HvKP,whose percentage was recently reported as rapidly elevated in 2013 by our team[33].Using the GPA package,we estimated that the rmpA gene was increased from 7 copies in 2009 to 13 copies in 2013.Also,we estimated that the copy number of rmpA2 was increased from 10 to 13.This increase was consistent with our observations in individual genomes and phenotypes,although the copy number was not predicted beforehand[33].tolC is a common protein subunit among many multidrug eff lux complexes in Gram-negative bacteria.This genewasobserved to have a partially deleted region of~90 bp in 10 strains in the2009 dataset,but only 2 strains in the 2013 dataset.Using the GPA package,we detected a deleted region in the gene in the 2009 data but failed to detect it in the2013 data(Figure5B).In a comparison of differences in estimated copy number between thetwo datasets,GPA also identif ied a signif icant increase of the strain without deletions in 2013,which suggested that potential antibiotic resistance might be elevated by the active eff lux of the drug from the periplasmic entrance using the eff lux complex(Table S2).The estimated result by GPA was also more stable than with DBE in our analysis of a single gene(Figure 5B).

Table 1 The discrimination of SNVs using GPA and GATK
We also estimated the antibiotic gene frequency in metagenomics data.The mex F gene,a resistance-nodulation-cell division(RND)multidrug eff lux transporter,was present in 0.6%deletion regionsin MH0001,in 65.7%of theduplication regions in MH0005,and conserved without mutation in MH0018.However,lacking of phenotypic and individual genomic data,we could not well evaluate our accuracy in this dataset.
Discussion
Metagenomics tools provide powerful insights into the study of populations of clinical or pathogenic microbiota[34].To fully exploit its potential in the discrimination of genetic variants,including CNVs and SNVs,data analysis tools and algorithms are needed for detecting dynamic changes in complex communities,and evaluating their effects on toxin production,antimicrobial resistance,and other phenotypes[27].The GPA package is a Bayesian approach for estimation of microbial genetics modal assignments,especially useful in complex clinical samples(Table S3).Using individual genomesand Pool-seq genomic data,we assessed the accuracy,sensitivity,and stability in a uniform model.Compared to the traditional method,this model eff iciently decreased the false discovery rate(FDR)of CNV and MAEof SNV in genotypecalling.Considering that SNV and CNV prof iles were two major factors in the analysis of metagenomic samples,and given the limited general methods that can estimate their distribution and relative abundance,we propose that Bayesian estimation could improve the discrimination of CNVs and SNVs between populations.In addition,we have constructed a GPA package with a complete workf low to analyze Pool-seq data from clinical or environmental samples,which will help researchers to automatically align reads to a selected reference,identify CNV and SNV variants,calculate the accuracy of the allele fraction,and annotate these variants.
In the GPA package,wehaveimproved threeaspectsof the genotyping of bacterial genomes.First,the GPA package uses the Bayesian model to detect CNV allele fraction,which decreases FDR and MAEsignif icantly compared to traditional DBE.Second,the SNV allele fraction is modif ied according to the CNV allele fraction,to correct the bias caused by the deletion allele.The accuracy of genotyping provides a foundation for further genotype-phenotype study.Third,this is a complete package containing themajor workf low used in microbial assignment in complex samples,and optimized with Pool-seq and whole genome/metagenomics sequence data from clinical settings,which could beused in hospital-based epidemiological molecular surveillance.
A limitation of GPA is the selection of the represented reference sequences.The integrity and complexity of the references may inf luence the statistical estimation of deletions,insertions,duplications,and translocations,which occur in bacterial populations.A sliding ploidy number used in the model will also extend the area of its application,instead of the assumed ploidy number given in our multi-ploidy model.For its application in metagenomics,we assume the sample is a multi-ploidy model,which may not be true in actual samples.The computing time increases exponentially with the increase in ploidy number,which also limits the application in metagenomics sequence data since we cannot increase the ploidy number too much to obtain an approximation in true metagenomics samples.Metagenomics and Pool-seq aremajor breakthrough technologies in clinical identif ication.Bayesian estimation provides an algorithm for accurate and thorough study of the epidemiology,distribution,and pathogenic potential of the reference strain.This approach will also provide considerable insight into other characteristics of individual strains,such as sequence type and gene evolution.
Materials and methods
Materials used for GPA evaluation
Wholegenomesequencing and antibiotic resistanceassaysfor 44 isolates of K.pneumoniae
We randomly selected 44 K.pneumoniae isolates obtained in 2009 and 2013(22 in each year)(Table S2).The genomic DNA from these isolates was extracted and purif ied using a QIAamp DNA Mini Kit(QIAGEN,Hilden,Germany).Then we prepared 500 bp libraries from the genomic DNA of each isolate using NEBNext Ultra DNA Library Prep Kit for Illumina.The libraries were sequenced on the Illumina Hiseq 2500 platform to generate 125 bp paired-end reads(Illumina,San Diego,CA,USA)according to the Illumina manual.The sequence data were deposited in the GenBank database(Accession number:SRP075790).The MICs of these isolates tested with 10 antibiotic agents from eight categories were determined using the broth microdilution method(Table S2).
Simulated data of individual genomesequencedata and Pool-seq data
We prepared simulated data to evaluate the accuracy of the GPA package for different coverage depths and ploidies by sampling the data from individual~150×shotgun reads.NC_016845 was used as reference genome.The BAM f iles were sampled with the DownsampleSam program in PICARD tools(v1.141).Each raw sample was down-sampled to 3 independent replicates with different coverage depths(5×,10×,15×,20×,50×,100×).For the given depth of each sample,we randomly selected the simulated reads from other bacteria to generated the pooled sequence data with 3,5,10,20 mixed sample data.
Evaluation of simulated data
We mapped the simulated and original data from FASTQ f iles to the reference genome using BWA-mem with default parameters,and then removed unmapped reads,low quality reads(Q<30),and multiple mapping reads to obtained the BAM f ile.These BAM f iles were used to calculate the sequencing depth and coverage for each genome.GPA and DBE approaches were evaluated using simulated Pool-seq data and individual genome shotgun sequence data.
Pool-seq data and metagenomics data
We mixed DNA from the aforementioned 22 K.pneumoniae isolates from both years using average concentrations to make a uniform level of DNA.We then constructed 2 Pool-seq libraries using NEBNext Ultra DNA Library Prep Kits for Illumina and then sequenced on an Illumina Hiseq 2500 platform.Metagenomics data were downloaded from the BGI website (http://gutmeta.genomics.org.cn/), with accession number from MH0001 to MH0020 according to previously described methods[27].These two datasets were also used to evaluate the discrimination of GPA in simulated data.
Description of the model for CNV detection
Likelihood ploidy state at each position is correct

For the Pool-seq data of n samples,we considered 2*n+1 ploidy states in this position.In each position i,the genotype θicould be expressed as The Poisson model has been widely employed for analyzing sequence data as well as for differential gene expression[35].However,the Poisson distribution assumes that the variance of reads is equal to its mean,and with only one parameter.The negative binomial model permitted additional parameters that could be used as an alternative to model the larger variance,for examplein CNV detection and Pool-seq data analysis[36].In the GPA model,wemodeled thedepth of each position by a negative binomial distribution,
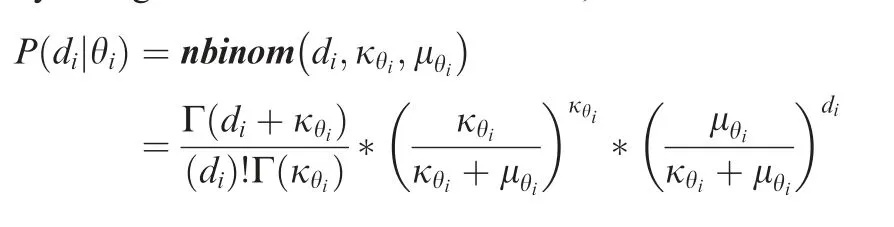
The dispersion parameterκθiwas set as a global parameter κfor each single experiment.The mean parameterμθiwas the expected depth of each site,which should be proportional to the ploidyθi,i.e.,μθi=a*θi.
We assumed that most positions were normal ploidy,which means that the main peak of depth distribution is contributed byθi=1.To cover more than 95%of the non-zero regions in sequencing depth,we def ined the range of 1/2 to 3/2 as the median depth.This peak value works well in single genome(Kpn24 genome as an example),3 pooled genomes(Kpn12,Kpn14,and Kpn32),and true metagenomics data(MH0005)in our study(Figure S4).We applied the maximum likelihood method to f it the main peak of observed depth distribution,estimated dispersion parameterκ,and the coeff icient a.The likelihood was calculated withμθi.Forθi=0,theμθiwas set as 1 since there are a few false positive reads in total deletion regions.
Iteratively updating the prior of the ploidy state
Prior probabilitiesof each ploidy stateswereestimated with an iterative algorithm.We started from equal probabilities,
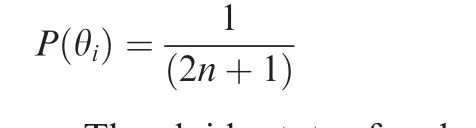
Theploidy stateof each position in thedata wasdetermined by the highest posterior probability,which was calculated asP(θi|di)∝P(di|θi)*P(θi)
After determining the ploidy state of each observed position,theprior probabilities of ploidy state P(θi)wereupdated.The repeated process will continueuntil P(θi)convergence to a stable state.The genotyping quality score was calculated as a Phred score of the maximum likelihood to qualify the conf idence of the results.
SNV allele fraction correction of the deletion genotyping data
For themajority of bacterial genomes,deletionsare widely distributed,potentially ashigh as 10%compared to thereference genome.The effect of CNVs cannot be ignored in SNV detection.For a particular position in the genome,ignoring a deletion would lead to an over-estimated allelecount of alternative alleles,while ignoring a duplication might lead to a decreased allele fraction.
In our pipeline,weapplied the Unif iedGenotyper in GATK software(version 3.4)[17]to detect bi-allelic SNVs.The parameter‘‘-ploidy”for each position was set according to the CNV result.
Mutation annotation and population analysis
Functional annotation of mutations is important in the research of a bacterial population.However,for the diversity of bacterial genomes,thereisnot a general method for a mutation annotation pipeline.Breseq is not f lexible in the annotation of a custom mutation list and genes.We have developed a pipelinefor mutation annotation based on ANNOVAR(version 2017-06-01)[32].ANNOVAR software is designed for human mutation annotation,and also provides solutions for novel genomes and gene annotations.A genome FASTA f ile and a gene annotation f ile in GenePred format were required for f ile preparation.Additional programs for f ile format conversion were provided in our software package.The AMR gene was annotated by the Resistance Gene Identif ier pipeline(RGI v3.1.1)using the Comprehensive Antibiotic Resistance Database(CARD v1.1.7)[31].
In the comparison mode,the GPA package provided information on the difference in allele frequency between two populations.Mutations with an allele frequency were listed in the results,with the statistical signif icance calculated by Fisher's exact test[37].
Authors’contributions
CC and HZ conceived the study.CC and JL designed the package.JL built the package and performed the statistical analysis.PD participated in the microbial gene annotation and population analysis.YY participated in the statistical model building.YZ and CScarried out the bacterial isolation,culturing and sequencing.CC and JL drafted the manuscript.All authors discussed the results,commented on the manuscript,and approved the f inal version for publication.
Competing interests.
The authors have declared no competing interests.
Acknowledgments
This work was supported by the Beijing Municipal Science&Technology Commission(Grant No.Z161100000516021),the National Key R&D Program of China(Grant No.2016YFC1200804),and the National Natural Science Foundation of China(Grant Nos.81571956 and 81702038).
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.gpb.2018.12.005.
 Genomics,Proteomics & Bioinformatics2019年1期
Genomics,Proteomics & Bioinformatics2019年1期
- Genomics,Proteomics & Bioinformatics的其它文章
- Acknowledgments to Reviewers 2018
- How Microbes Shape Their Communities A Microbial Community Model Based on Functional Genes
- Agricultural Risk Factors Inf luence Microbial Ecology in Honghu Lake
- Inulin Can Alleviate Metabolism Disorders in ob/ob Miceby Partially Restoring Leptinrelated Pathways Mediated by Gut Microbiota
- Eあects of Proton Pump Inhibitors on the Gastrointestinal Microbiota in Gastroesophageal Ref lux Disease
- Antibiotic Treatment Drives the Diversif ication of the Human Gut Resistome