
Agricultural Risk Factors Inf luence Microbial Ecology in Honghu Lake
2019-05-14 10:07:28MaozhenHanMelissaDsouzaChunyuZhouHongjunLi
Maozhen Han ,Melissa Dsouza ,Chunyu Zhou ,Hongjun Li,
Junqian Zhang 6,e,Chaoyun Chen 1,f,Qi Yao 1,g,Chaofang Zhong 1,h,Hao Zhou 1,i,Jack A Gilbert 3,4,5,*,j,Zhi Wang 2,*,k,Kang Ning 1,*,l
1 Key Laboratory of Molecular Biophysics of the Ministry of Education,Hubei Key Laboratory of Bioinformatics and Molecularimaging,Department of Bioinformatics and Systems Biology,College of Life Science and Technology,Huazhong University of Science and Technology,Wuhan 430074,China
2 Key Laboratory for Environment and Disaster Monitoring and Evaluation of Hubei,Institute of Geodesy and Geophysics,Chinese Academy of Sciences,Wuhan 430077,China
3 The Microbiome Center,Department of Surgery,University of Chicago,Chicago,IL 60637,USA
4 Argonne National Laboratory,Biosciences Division,Lemont,IL 60439,USA
5 Marine Biological Laboratory,Woods Hole,MA 02543,USA
6 State Key Laboratory of Water Ecology and Biotechnology,Institute of Hydrobiology,Chinese Academy of Sciences,Wuhan 430072,China
KEYWORDS Freshwater;Microbial communities;Agriculture activities;Antibiotics;Human impact
Abstract Agricultural activities,including stock-farming,planting industry,and f ish aquaculture,can affect the physicochemical and biological characters of freshwater lakes.However,theeffects of pollution producing by agricultural activities on microbial ecosystem of lakes remain unclear.Hence,in this work,we selected Honghu Lake as a typical lake that is inf luenced by agriculture activities.We collected water and sediment samples from 18 sites,which span a wide range of areas from impacted and less-impacted areas.We performed a geospatial analysis on the composition of microbial communities associated with physicochemical properties and antibiotic pollution of samples.The co-occurrence networks of water and sediment were also built and analyzed.Our results showed that the microbial communities of impacted and less-impacted samples of water were largely driven by the concentrations of TN,TP,NO3--N,and NO2--N,while those of sediment were affected by theconcentrationsof Sed-OM and Sed-TN.Antibiotics havealso played important roles in shaping these microbial communities:the concentrations of oxytetracycline and tetracycline clearly ref lected the variance in taxonomic diversity and predicted functional diversity between impacted and less-impacted sites in water and sediment samples,respectively.Furthermore,for samples from both water and sediment,large differences of network topology structures between impacted and less-impacted were also observed.Our results provide compelling evidence that the microbial community can beused asa sentinel of eutrophication and antibioticspollution risk associated with agricultural activity;and that proper monitoring of thisenvironment is vital to maintain a sustainable environment in Honghu Lake.
Introduction
Water ecosystems,especially inland lakes,have suffered from eutrophication associated with increased agricultural activity comprising f ish aquacultureaswell ascrop and livestock farming on surrounding lands[1-3].Improperly managed agricultural activities,such as excessive and/or improper use of fertilizers and/or pesticides,can cause eutrophication,which can negatively impact biodiversity[4].Previous studies have reported that the effect of this pollution on macroorganismal communities[4,5]and in comparison,microbial ecology remains relatively understudied.
Agricultural pollution alters the physicochemical properties of water ecosystems[6]and changesthe composition of microbial community.In particular,nitrogen and phosphorus content,water temperature,and pH can fundamentally inf luence the microbiome[7-9].However,few studies have quantif ied the impact of organic pollutants such as herbicides and antibiotics.Determining the ecosystems resilience to such disturbance can aid conservation and help in the development of remediation strategies.There is an urgent need to develop sustainable approaches that establish a balanced relationship between the environment and agricultural production.
Antibiotics are widely utilized in livestock and f ish aquaculture to promote animal growth and for the prophylactic or curative treatment of infectious disease[10],yet surface runoff of the introduction of treated sewage can introduce antibiotic pollution into local water bodies.Antibiotics inhibit microbial activity and can therefore inf luence biogeochemical processes in these ecosystems[11]and potentially select for antibiotic resistance mechanisms in environmental bacteria[12].In addition,animal sewage can introduce animal-associated antibiotic resistant bacteria into these environments[13],and as such it is necessary to have better quantif ication of the f itness and recovery rates of these resistant microbes upon release into the environment[14].
Honghu Lakeisa largeand a shallow eutrophic lake,which islocated between the irrigation channel of the Four-lake main canal and the Yangzi River.Its area is about 350 km2with an average depth of~1.5 m(Figure 1).In the last f ive decades,Honghu Lake has been extensively altered by f lood regulation,irrigation,f ish aquaculture,shipping,and water supply demands[15,16].Today,more than 40%of the lake area is used for large-scale aquaculture[17].The intensive useof Honghu lake resources and the emission of sewage and other pollutants including fertilizers,pesticides,and antibiotics into the lake have led to a severe degradation of its water quality and an increase in the frequency of eutrophication events.In 2004,the Honghu Lake Wetland Protection and Restoration Demonstration Project[17]was implemented to ameliorate the negative effects of severe water pollution,and one third of the lake area has been gradually protected under this provision.Consequently,Honghu Lake represents a valuable,natural f ield site for investigating both the eff icacy of the restoration program and the long-term effects of agricultural activities,such as the excessive application of antibiotics on water microbial communities.
This study aimed to understand the geospatial inf luence of pollution on thewater and sediment-associated microbial communitiesin Honghu Lake.Weperformed 16SrRNA amplicon sequencing to characterize the microbial ecology,which was correlated with physicochemistry and antibiotic concentrations in theseenvironments.Thisresearch wasguided by the following scientif ic questions:(i)How does microbial diversity differ between water and sediment?(ii)How does microbial diversity differ between less-impacted and impacted samplesin water and sediment,respectively?(iii)Which physicochemical properties and antibiotics are correlated with changes in microbial community structure?(iv)How aretheco-occurrencerelationships between microbiota inf luenced by the intensity of agricultural pollution(impacted and less-impacted)?Importantly,thisbaseline study aims to generate microbial biomarkers of pollution,and to identify the ecological trends that could be used to provide a sentinel of pollution events in this lake environment.
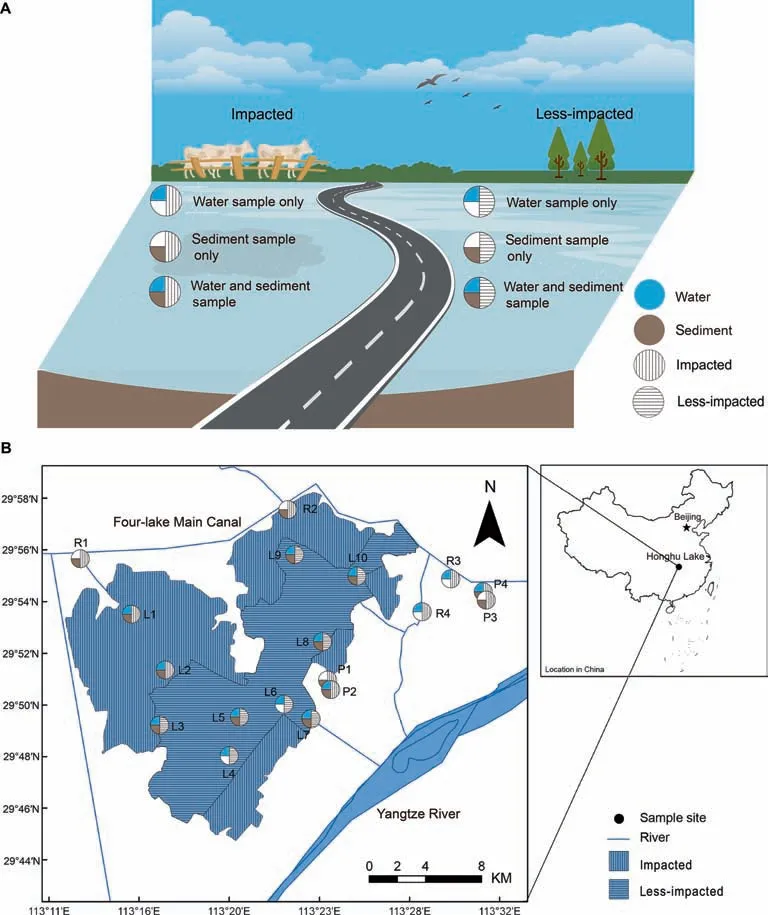
Figure 1 Geographic distribution of all sampling sites in Honghu LakeA.Def inition of various sampling strategies.B.Locations of sampling sites and thedistribution of thesampling medium collected at each site.Theshaded segment areaswith horizontal linesand vertical linesrepresent theless-impacted sitesand impacted sites in Honghu Lake,respectively.Sites are named according to location of the sampling.L,lake;P,pond;R,river.
Results
Physicochemical and antibiotic characterization
Physicochemical characteristics and antibiotic concentrations were determined for all water and sediment samples(Figure 1,File S1,Tables S1,S2,and S3).Signif icant differencesin p H(ttest,P=0.0037),oxidation-reduction potential(ORP,t-test,P=0.00068),total nitrogen(TN,t-test,P=0.035),and ammonium nitrogen (NH4+-N,t-test,P=0.045)were observed between water samples from impacted and lessimpacted(control)sites(Table S1).Samples from impacted sites were signif icantly more acidic and had greater concentrations of ORP,TN,and NH4+-N when compared to less-impacted sites(Table S1).Similarly,sediment samplesmaintained signif icantly different sediment organic matter(Sed-OM,t-test,P=0.002),sediment labile phosphorus(Sed-LP,t-test,P=0.0335), sediment total nitrogen (Sed-TN, t-test,P=0.0013),and sediment total phosphorus(Sed-TP,t-test,P=0.021)levels between impacted and less-impacted sites(Table S2).Less-impacted sediment had greater concentrations of Sed-OM,Sed-LP,and Sed-TN when compared to the impacted sites(Table S2),which may largely be due to the decomposition of plant material over the preceding winter months.Between impacted and less-impacted sites,the antibiotics of loxacin (OFL, t-test, P=0.0079) and sulfamethoxazole(SMZ,t-test,P=0.043)had signif icantly different concentrations in water samples,while sulfamerazine(SMR,t-test,P=0.021)was signif icantly different in sediment samples(Table S3);in both cases concentrations were greater in impacted sites.
Microbial diversity and community structure
A total of 28 water and sediment samples generated 4,441,405 paired-end 16SrRNA reads,which clustered into 7785 OTUs(File S1,Table S4).Microbial alpha diversity was signif icantly greater in sediment samples[Chao1 (t-test,P=0.0045,Table S4)and phylogenetic diversity(PD)whole tree(t-test,P=0.003,Table S4)].The microbial alpha diversity in sediment samples was signif icantly different between impacted and less-impacted sites(t-test,P=0.0445,Table S4).However,we observed that the alpha diversities in water samples have no signif icant differences.
A total of 53 microbial phyla wereidentif ied acrossall samples(Figure 2A),and were differentiated between water and sediment samples(Figure 2B),and between impacted and less-impacted sites(PERMANOVA,Bray-Curtis distance,P<0.01). In water samples, Proteobacteria (t-test,P<0.05),Cyanobacteria(t-test,P<0.05),and Gemmatimonadetes (t-test,P<0.05)were signif icantly different between impacted and less-impacted sites(Figure 2C).While in sediment samples,Actinobacteria(t-test,P<0.01),Firmicutes(t-test,P<0.05),Bacteroidetes(t-test,P<0.05),Nitrospirae(t-test,P<0.05),and OP8(t-test,P<0.05)were signif icantly different between impacted and less-impacted sites(Figure 2D).
Core-OTUs were def ined as a set of OTUs that were identif ied in all samples analyzed,and pan-OTUs were def ined as a set of OTUs that were identif ied in at least one sample.Coreand pan-OTUs were determined for all water and sediment samples(Table S5,Figure 3).A total of 132 core-OTUs and 7418 pan-OTUs were identif ied in less-impacted sites,while impacted sites maintained 201 core-OTUs and 7706 pan-OTUs(Figures S1 and S2).The core-OTUs from both the impacted and less-impacted sites were dominated by Proteobacteria,specif ically Janthinobacterium (Tables S6 and S7),while Acidobacteria were enriched at the impacted sites(2.79%±1.30%,Table S6).
Microbial beta diversity was further assessed by Unweighted Pair Group Method with Arithmetic Mean(UPGMA)clustering using the unweighted UniFrac distance matrix.We observed clustering by sampling medium(Figure 4A and Figure S3)and by level of agricultural activity within water and sediment samples(Figure 4B).Importantly,greater differences in beta diversity were observed between impacted and less-impacted sites in sediment samples as compared to water samples(Figure 4B and C).

Figure 2 Taxonomic composition and relative abundances of microbial taxa in water and sediment samplesA.Taxonomic composition of each sample at the phylum level.‘Other'represents all phyla not included in the top 13 phyla.Samples are named according to thesampling sites(see Figure1B)with postf ixes Wand Sfor water and sediment,respectively.B.Bar plot highlighting differences between water and sediment samples at the phylum level.C.Bar plot highlighting differences in water samples at the phylum level between impacted and less-impacted groups.D.Bar plot highlighting differences in sediment samples at the phylum level between impacted and less-impacted groups.Student's t-test is performed to determine signif icant differences between samples collected from different sampling media(water vs.sediment)or locations(impacted and less-impacted).*P<0.05;**P<0.01.

Figure 3 Core-OTUs and pan-OTUs of water and sediment samples from Honghu LakeFlower plots showing the number of sample-specif ic OTUs(in the petals)and core-OTUs(in the center)for all samples(A),all water samples(B),and all sediment samples(C).OTU accumulation curvesfor pan-OTUs(top)and core-OTUs(bottom)for all samples(D),all water samples(E),and all sediment samples(F),respectively,from Honghu Lake.Samples are named according to the sampling sites(see Figure 1B)with postf ixes W and Sfor water and sediment,respectively.
Comparison of functional properties between less-impacted and impacted groups
We observed clustering of water and sediment microbial communities based on the relative abundance of their predicted functional prof iles(Figure S4)(PERMANOVA,Bray-Curtis distance,P<0.0001).In water samples,functional groups including amino acid related enzymes,peptidases,oxidative phosphorylation,purine metabolism,pyrimidine metabolism,DNA repair and recombination proteins,and arginine and proline metabolism were enriched(Figure S5).Likewise,we observed an enrichment of functional groups including ribosome biogenesis,secretion system,two-component system,ABC transporters,and pyruvate metabolism in sediment samples(Figure S5).When investigating agricultural pollution risks,we observed signif icant differences in the relative abundances of the predicted functional prof iles between impacted and less-impacted groups of water samples(PERMANOVA,Bray-Curtis distance,P<0.05).For these samples,the relative abundances of DNA repair and recombination proteins(t-test,P<0.05),purine metabolism(t-test,P<0.05),secretion systems(t-test,P<0.05),oxidativephosphorylation(t-test,P<0.05),pyrimidine metabolism(t-test,P<0.05),amino acid related enzymes(t-test,P<0.05),and arginine and proline metabolism(t-test,P<0.05)were signif icantly different between impacted and less-impacted sites(Figure S5).In contrast,we observed no signif icant differences in sediment functional prof iles between impacted and less-impacted sites.
Correlating physicochemical properties with microbial diversity
Physicochemical properties including NH4+-N,TN,ORP,TP,turbidity(Tur),potassium permanganate index(oxygen consumption,CODMn),and chlorophyll-a(Chl-a,Table S8)were signif icant explanatory factors that determined the observed clustering pattern of the water microbial communities at impacted sites(Figure 5A,Figure S6A),while p H and dissolved oxygen(DO)determined the water microbial community structure at less-impacted sites(Figure 5A,Figure S6A).For sediment samples,Sed-LP,Sed-TN,and Sed-OM(Table S9)were identif ied as signif icant explanatory factors shaping the observed clustering pattern at less-impacted sites and Sed-TPfor impacted sites(Figure 5B,Figure S6B).Based on distance correlations and thestatistical signif icanceof Mantel's r-statistic,water physicochemical properties including TN,ORP,nitrate nitrogen(NO3--N),and nitrite nitrogen(NO2--N),were strongly correlated with taxonomic and functional composition(Figure 6A).For sediment samples,Sed-OM and Sed-TN were strongly correlated with taxonomic composition(Figure 6B).
The antibiotic oxytetracycline(OTC)was the primarily explanatory factor for water microbial diversity variance at impacted sites(Figure 5C,Figure S6A).While in sediment samples,SMR was the primary factor responsible for the observed clustering of samples,including R1S,R2S,P1S,P3S,and P4S,from impacted sites(Figure 5D,Figure S6B).Mantel's correlation assessments were also performed between antibiotic data and compositional data for water and sediment samples(Figure 6C,D,and Figure S7).The OTC antibiotic class was strongly correlated with water taxonomic and functional composition(Figure 6C),while tetracycline(TC)was strongly correlated with sediment taxonomic and functional composition(Figure 6D).The OFL antibiotic class was strongly correlated with taxonomic and functional composition in sediment samples collected from less-impacted(control) sites (Figure S8B). Additionally, CODMnand ciprof loxacin(CIP)were strongly correlated with taxonomic and functional composition in water samples collected from impacted sites(Figure S8C).
Moreover,we observed strong correlations between several OTUs,physicochemical properties,and antibiotic concentrations(Table S10,Figures S9-12).In water samples,Bacillus f lexus(denovo 71031,Table S10)was strongly correlated with TN(r=0.8675,fdr-P=7.89E-5,Figure S9C),NH4+-N(r=0.8958,fdr-P=7.89E-5,Figure S9D),orthophosphate(PO43--P,r=0.832,fdr-P=2.58E-4,Figure S9E),and OTC(r=0.8381,fdr-P=3.62E-4,Figure S10B).
Biomarker discoveryIn water samples,the LEfSe analysis identif ied 13 biomarkers for impacted sites and 12 for less-impacted sites.The most differentially abundant bacteria from impacted sites belonged to the phylum Proteobacteria,class Betaproteobacteria and class Gammaproteobacteria(Figure 7A and B).These included members of the orders Methylophilales,
Nitrosomonadales,and Rhodocyclales(Figure 7A and B).Methylophilales are known for their ability to metabolize methane under aerobic and microaerobic conditions[18]and Nitrosomonadales are signif icantly enriched in soils containing high concentrations of N fertilizer[19].Water samples from less-impacted sites were overrepresented by Oscillatoriophycideae and Synechococcophycideae in Cyanobacteria; and Saprospiraceae in Bacteroidetes(Figure 7A and B).
In sediment samples,the LEf Se analysis reported 14 biomarkers enriched in impacted sites and 5 enriched in lessimpacted sites(Figure 7C and D).Biomarkers in samples from impacted sites mainly comprised members of the phylum Actinobacteria,family Pseudomonadaceae,order Burkholderiales,and class Flavobacteriia.For sediment samples from lessimpacted sites,bacteria that were differentially abundant include members of Paenisporosarcina genus and candidate family planococcaceae,phylum Firmicutes,order Bacillales,and class Bacilli(Figure 7C and D).
Co-occurrence network analysis

Figure 4 PCoA plots and UPGMA-based clustering of water and sediment microbial communitiesUnweighted UniFrac dissimilarity matrix scores for all samples were visualized in a PCoA plot to demonstrate the dissimilarity of the microbial community structurebetween samplesby samplingmedium,water vs.sediment(A)and by sampling location,impacted and lessimpacted(B).C.UPGMA-based clustering tree of microbial communities using an unweighted UniFrac distance matrix.The green and pink fontsrepresent less-impacted and impacted groups,respectively.Theblueand red barsmark water and sediment samples,respectively.Samples are named according to the sampling sites(see Figure 1B)with postf ixes W and Sfor water and sediment,respectively.
Co-occurrence network analysis was performed to visualize and characterize co-occurrence patterns among members of water and sediment microbial communities.The water and sediment network comprised 427 nodes and 189 edges(Figure 8A)and 443 nodesand 2877 edges(Figure8B),respectively.The density of the water and sediment network was 0.002 and 0.023,respectively.These results suggest that the sediment microbial network was more connected than the water network.Both networks exhibited a scale-free degree distribution pattern,whereby most OTUs had low degree values and fewer hub nodes had high degree values(Figure S13).

Figure 5 Canonical correspondence analysis plots of physicochemical properties and antibiotic data driving water and sediment microbial community structurePhysicochemical properties of water samples(A)and sediment samples(B),as well as antibiotic data for water samples(C)and sediment samples(D)from Honghu Lake.We utilized the‘envf it'function with 999 permutations to reveal signif icant correlations between physicochemical properties,antibiotics,and microbial communities.*P<0.05;**P<0.01;***P<0.001.DO,dissolved oxygen;ORP,oxidation-reduction potential;Tur,turbidity;Chl-a,chlorophyll-a;TP,total phosphorus;TN,total nitrogen;CODMn,oxygen consumption;OM,organic matter;LP,labile phosphorus;OTC,oxytetracycline;TC,tetracycline;SMR,sulfamerazine.
Wedetected modulesin water and sediment networksusing the Walk Trap community detection algorithm.The modularity of the water and sediment network was 0.878 and 0.559,respectively.A total of 50 clusters with the largest membership of 22 was observed for the water network(Figure 8C).Likewise,for the sediment network we observed a total of 38 clusters with the largest membership of 111(Figure 8D).In the sediment network,most OTUs in module 6(111 nodes)were members of Anaerolineae of the phylum Chlorof lexi and Beta-,Delta-,and Gammaproteobacteria.Additionally,most OTUs in module 7(81 nodes)of the sediment network were members of the Planococcaceae,a family within the order Bacillales.When compared to the sediment network,we observed fewer and smaller hubs in the water network.In this network,most OTUs in module 2(22 nodes)and module 9(12 nodes)were members of the genus Synechococcus within the order Synechococcales and ACK-M 1 within the order Actinomycetales,respectively.
Wealso examined theeffect of prolonged agricultural activities on the patterns of co-occurrence network from water and sediment microbial communities,respectively.For this,each nodein thewater and sediment network wascolored asa function of its relative abundance across samples from impacted and less-impacted(control)sites(Figure 8E and F).In both networks,we observed higher connectedness among OTUs associated with less-impacted samples as compared to those associated with samples from impacted sites.We conf irmed this observation in impacted and less-impacted sediment samples by selecting OTUs that related to these sediment samples and all its edges from the overall sediment co-occurrence network to generate the sub-networks(Figure S14).We observed higher connectedness in microbes associated with lessimpacted samples(measured as node degree,3.746)as compared to those associated with samples from impacted sites(1.397).

Figure 6 Environmental drivers of microbial community composition in water and sediment samplesPairwise comparison of physicochemical properties with taxonomic and functional composition data in water(A)and sediment(B)samples.Pairwise comparisons of antibiotic concentration data with taxonomic and functional composition data in water(C)and sediment(D)samples.The actual PCC values are indicated in color gradient(with green for lower PCC values and red for higher PCC values),while theabsolute PCC valuesareindicated using circle with bigger sizerepresenting higher absolute PCC values between thetwo factors.The edge width represents Mantel's R statistic value for distance correlation and the edge color denotes the statistical signif icance(P values)based on 9999 permutations.PCC,Pearson's correlation coeff icient;nlF Cond,temperature compensated conductivity;Sal,salinity;DO,dissolved oxygen;ORP,oxidation-reduction potential;Tur,turbidity;Chl-a,chlorophyll-a;fDOM,f luorescent dissolved organic matter;TP,total phosphorus;TN,total nitrogen;CODMn,oxygen consumption;OM,organic matter;LP,labilephosphorus;TC,tetracycline;OTC,oxytetracycline;CTC,chlortetracycline;SDZ,sulfadiazine;SMR,sulfamerazine;SMD,sulfadimidine;OFL,of loxacin;CIP,ciprof loxacin;SMZ,sulfamethoxazole.
Discussions
The extensive application of chemical compounds such as fertilizers,herbicides,and antibiotics,can profoundly inf luence the cycling and accumulation of nutrients in the sediment and water column of Honghu Lake[20].These agricultural practices can negatively impact not only the physicochemical properties,but also the biodiversity of microbial communities associated with the lake ecosystem[6].These changes in microbial community composition can in turn affect nutrient cycling and organic matter decomposition,thus impacting overall agricultural productivity.
In our study,we analyzed water and sediment samples from Honghu Lake,assessing its microbiome,physicochemical properties,and antibiotic concentrations.We found that despite low human activity,high concentrations of Sed-LP,Sed-TN,and Sed-OM wereobserved at less-impacted(control)sites,probably due to the abundance of submerged plants.We speculate that the decay of these plants during winter substantially increases organic matter,total nitrogen[21],and total phosphorus[22]in sediment samples.Hence,as expected from previous research,we found that both water and sediment microbial community structure was correlated with TP and TN concentration[23,24].Moreover,in water samples,we observed that Bacillus f lexus was strongly correlated with TN,NH4+-N,PO43--P,and oxytetracycline.More important,previous work on Bacillus f lexus have shown that members of this species can degrade organic[25]and inorganic[26]nitrogen,thus making it a possible candidate for bioremediation in alkaline wastewater[27].Some strains of B.f lexus also demonstrate strong phosphorus solubilization activity[28],and others demonstrated resistance to OTC[29].

Figure 8 Co-occurrence network interactions of Honghu Lake microbes in water and sediment samplesNetwork nodes represent OTUs with the size of each node proportional to the node degree.Edges represent positive,strong(Spearman's ρ>0.8),and signif icant(P<0.001)interactions between OTUs.Networks of water(A)and sediment(B)samples displaying cooccurrencepatternsof OTUsgrouped at thephylum level.Moduleswereidentif ied using the WalkTrap community detection algorithm in water(C)and sediment(D)samples.Networks of water(E)and sediment(F)samples investigating the effect of long-term agricultural activitieson microbial community wherein each nodeiscolored asa function of itsrelativeabundanceat impacted and less-impacted sites.
As to biomarkers in sediment samples from impacted sites,these included members of the Hydrogenophaga genus,belonging to Burkholderiales (Class Betaproteobacteria),which have been previously associated with agricultural activities[30].Moreover,members of the genus Pseudomonas,belonging to family Pseudomonadaceae,can play an important role in agricultural ecosystems,particularly those associated with plant growth-promotion and disease suppression were mentioned[31].
Co-occurrence network analysis showed that Anaerolineae forms a large component of microbial communities associated with sludgewastewater treatment plantswherein they may play important roles in organic degradation[32].The phylum Proteobacteria areknown to easily metabolizesolubleorganic substrates[33].Among these classes,Deltaproteobacteria,a dominant group often observed in various sediment samples,play an important rolein degrading organic compoundsto carbon dioxide[34].Membersof Synechococcus areacosmopolitan cyanobacterium often associated with toxic algal blooms and microcystin production[35,36].Likewise,members of ACKM 1,in a recent study,exhibited chemotaxistoward ammonium in awater ecosystem,thusinf luencingnutrient cyclingprocesses and microbial competitive interactions within this ecosystem[37].The presence of these microbial taxa is indicative of the long-term effect of eutrophication in water environments.
Conclusion
We analyzed the impacted sites and less-impacted sites of water and sediment samplesfrom Honghu Lakeand surrounding river and pond sites.The microbiome was analyzed in the context of variable physicochemical properties and antibiotic concentrations.There were signif icant differences between impacted and less-impacted(control)groups in both water and sediment samples.These differences were observed in physicochemical properties,antibiotic concentration levels,and taxonomical structure.Physicochemical properties including TN,TP,NO3--N,and NO2--N were the main factors driving compositional differences in water samples.Likewise,in sediment samples,Sed-OM and Sed-TN were the main factors driving differences in taxonomical composition.The antibiotics,oxytetracycline and tetracycline were identif ied as the main drivers of taxonomical and functional structure in water and sediment samples,respectively.As for differences between impacted and less-impacted samples,weidentif ied 25 biomarkers within water microbial communities and 19 within sediment microbial communities.Finally,the co-occurrence network analysisrevealed differencesin co-occurrencepatterns by sampling medium(water vs.sediment microbial communities)and by level of agricultural activity(impacted vs.lessimpacted microbial communities).These results suggest that continued analyses of the composition and structure of water and sediment microbial communities in such anthropologicallyimpacted lake environments may provide valuable biomarker data to track pollution.The Honghu Lake Wetland Protection and Restoration Demonstration Project provided preliminary data that highlights the importance of monitoring biodiversity in water micro-ecosystems.Our present work allows further investigation into the impact of agricultural practices on water ecosystems and more importantly,into our ability to remediate these important ecosystems.
Materials and methods
Sample sites and sampling processes
To investigate the differences in microbial community structure resulting from a wide range of anthropogenic activities,a total of 14 water samples and 14 sediment samples were collected from Honghu Lake and surrounding rivers and ponds during 10-11 November 2015.Among these sites,site L1 is the entrance of inf lowing river,and sites L3,L8,L9,and L10 are relatively adjacent to aquaculture district.Meanwhile,to evaluatethemain sourceof theantibioticsof Honghu Lake,sites R1,R2,R3,and R4,which are located in four major connecting rivers of Honghu Lake and sites P1 to P4,four typical aquaculture ponds,which can swap water with Honghu Lake,were collected[38].In keeping with the Government Protection Zone def inition[17]and in taking into account the different sources of pollution at each site[38](treated sewage,crop,livestock,and f ish aquaculture),all sampling sites were categorized into two groups—namely,the impacted and the lessimpacted(control)groups[17].Sampling sites labeled L1,L2,P1,P2,P3,P4,R1,R2,R3,and R4 were classif ied as impacted,while sites labeled L3,L4,L5,L6,L7,L8,L9,and L10 were classif ied as less-impacted(Figure 1).
For water sampling,2 L of water with a depth of 0.3-0.5 m were collected at each sampling site using a cylinder sampler.Approximately 1.5 L of sample was used for physicochemical characterization and antibiotic analysis.The remaining 500 mL of sample was size-fractionated using a 20μm tulle and a 0.22μm diameter pore size f ilter membrane(Tianjin Jinteng Experiment Equipment Co.,Ltd).Microbial biomass was collected on 0.22μm diameter pore size f ilter membranes.These membrane samples were stored onsite in a portable cooler with ice bags,then transported to the laboratory and stored at-80°C until DNA extraction.For sediment sampling,~200 g of sediment(0-10 cm)was collected at each site and stored in a portable cooler with ice bags until its transportation to the laboratory for subsequent downstream analyses.Approximately 50 g of sediment was used for physicochemical characterization and antibiotic analysis,while the remainder was dried in an Ultra-low Freeze Dryer(Christ,German)until no further weight changes were observed.The dried sediment(0.5 g)was used for DNA extraction.
Physicochemical characterization and antibiotic analysis
Physicochemical characterization
Physicochemical data were measured for all water and sediment samples(Tables S1 and S2).Physicochemical properties including water temperature(T),p H,temperature compensated conductivity(nlF Cond),DO,salinity(Sal),ORP,Tur,Chl-a,and f luorescent dissolved organic matter(f DOM)were measured for all water samples in situ by EXO2(YSI).Additional physicochemical properties including TP,TN,NH4+-N,CODMn,PO43--P,NO2--N,and NO3--N were assayed as described in previous work[39].For sediment samples,ORP(Sed-ORP)and pH(Sed-pH)were determined using a p H/ORP portable meter(YSI).Sed-OM was determined in a muff le furnace at 550°C[39].Sed-LP,Sed-TP,NH4+-N(Sed-NH4+-N),and Sed-TN were measured by the NH4Clextraction method,the KCl extraction method,the perchloric acid and sulfuric acid digestion method,and the Kjeldahl method,respectively[40].
Antibiotic analysis
Based on a report of antibiotic usage in China[41],a total of 13 antibioticswere selected for detection in water and sediment samples(Table S3).These antibiotics were classif ied into three groups namely:(i)sulfonamides(SAs),including sulfadiazine(SDZ),SMR,sulfamate(SFM),sulfadimidine(SMD),sulfamonomethoxine(SMM),and SMZ;(ii)f luoroquinolones(FQs),including f leroxacin(FLE),OFL,CIP,and dif loxacin(DIF);and(iii)the tetracycline group(TCs),including TC,OTC,and chlortetracycline(CTC).Wedetermined theconcentration of these antibiotics in water and sediment samples using a 2695 Waters Alliance system(Milford,MA).A detailed protocol of the antibiotic extraction process was described in File S1.Of the 13 antibiotics that were quantif ied,nine antibiotics including TC,OTC,CTC,SDZ,SMR,SMD,OFL,CIP,and SMZ were selected for further analysis in this study.
DNA extraction and 16S rRNA gene sequencing
DNA was extracted from water f ilter membranes and dried sediment using a modif ied hexadecyltrimethylammonium bromide(CTAB)method[42-44].All extracted metagenomic DNA was dissolved in TE buffer and stored at-20°C until further use.
Metagenomic DNA were quantif ied by using a Qubit®2.0 Fluorometer(Invitrogen,Carlsbad,CA)and the quality of DNA was assessed on 0.8%agarose gels.Approximately 5-50 ng of DNA was used as template for amplifying the V4-V5 hypervariable region of the 16Sr RNA gene of microbiota for each sample.Sequences for the paired primers are‘‘GTGYCAGCMGCCGCGGTAA”and‘‘CTTGTGCGGK CCCCCGYCAATTC”,respectively[24].The sequencing library was constructed using a MetaVxTMLibrary Preparation kit(GENEWIZ,Inc.,South Plainf ield,and NJ).The ends of the 16S r DNA amplicons were added with indexed adapters by limited cycle PCR.Sequencing libraries were verif ied using the Agilent 2100 Bioanalyzer(Agilent Technologies,Palo Alto,CA)and quantif ied by Qubit®2.0 and quantitative PCR(Applied Biosystems,Carlsbad,CA).All amplicons were sequenced on the Illumina MiSeq platform (paired-end,2*300 bp).All sequencing data for the 14 water samples and the 14 sediment samples were deposited into NCBI's Sequence Read Archive(SRA)database under the Bioproject number PRJNA352457.
Quality control,OTU clustering,and taxonomy assignment
All 16SrRNA geneampliconswereprocessed according to the ensuing criteria and sequences below the set quality threshold were excluded from subsequent analyses.Firstly,paired-end reads were spliced using the‘make.contigs'command in mothur[45](version 1.25.0)with default settings.We conducted the quality control to remove the low-quality reads,which contained ambiguous base calls(N),or longer than 500 bp,and those shorter than 300 bp.Putative chimeras were identif ied using the SILVA database[46](Release 123)and removed using the‘chimera.uchime'and‘remove.seqs'commands in mothur.All high-quality sequences were aligned using PyNAST and dereplicated with UCLUST[47]in QIIME(Quantitative Insights Into Microbial Ecology,v1.9.1)[48].Finally,the Greengenes database(version 13_8)[49]was used as the reference database for classifying de novo operational taxonomic units(OTUs)that were clustered with the 97%nucleotide identity.We set 0.001%as the threshold to f ilter the low-abundance OTUs and keep abundant OTUs for analysis[50].
Microbial diversity assessment
Microbial alpha-and beta-diversity values were determined using the QIIME[48]pipeline.For alpha-diversity,rarefaction curves were drawn based on the following metrics:Observed OTUs,Chao1,PD whole tree metric,and the Shannon evenness metric[51].For beta-diversity analysis,the f inal OTU table was raref ied to contain 61,088 reads per sample.Bray-Curtis,weighted and unweighted UniFrac distance metrics[52]were used to measure community similarity among samples.Microbial community clustering was arrayed by Principle Coordinates Analysis(PCoA)and visualized using Emperor[53]in QIIME.The hierarchical clustering method,UPGMA,was applied to cluster all water and sediment samples,and the clustering tree was visualized in FigTree(version 1.4.2,http://tree.bio.ed.ac.uk/software/f igtree/).Permutational multivariate analysis of variance(PERMANOVA)[54]was performed on the Bray-Curtis distance matrix to compare differences in community structure.
Functional prof iling
PICRUSt(version 1.0.0-dev)[55]was used to make functional predictions based on the 16SrDNA dataset from each sample.For this,OTU-picking was performed on all quality-f iltered sequence data using the‘pick_closed_reference_otus.py'command in QIIME.OTUs were clustered at the 97%nucleotide identity threshold using the Greengenes database.The OTU table was normalized using the‘normalize_by_copy_number.py'command.The normalized OTU table was used for functional prediction with the‘predict_metagenomes.py'script,and functional trait abundancesweredetermined for each sample using the KEGG database(version 66.1,May 1,2013)[56].Finally,the predicted functional content was collapsed to level three of the KEGG hierarchy using the‘categorize_by_func tion.py'script.
Analysis of the relationships between physicochemical properties,antibiotics,and microbial communities
Canonical correspondence analysis(CCA)was chosen and used to identify an environmental basisfor community ordination,revealing relationships between microbial communities and environmental factors[57].For this,the CCA function in R package,vegan was utilized.We utilized the‘envf it'function[58,59]with 999 permutations to reveal signif icant correlations between physicochemical properties,antibiotics,and microbial communities.To further investigate correlations between environmental factors(including physicochemical properties and antibiotics) and OTUs,we applied a low-abundance f ilter to remove OTUs whose relative abundance did not exceed 0.01%in any sample(as previously reported by[60]).Similarly,for physicochemical data and antibiotics data,the values of each variable were transformed to z-scores[61],based on which the Pearson Correlation Coeff icient between each environmental factor and each OTU was calculated.To select for signif icant interactions between an environmental factor and an OTU,the threshold of the r-value and the False Discovery Rate(FDR)-corrected P value of the Pearson Correlation Coeff icient was set at 0.8 and 0.05,respectively.
Analysis of environmental drivers of microbial community composition
We noted environmental drivers of microbial community composition on the basis of(i)compositional data,which include taxonomical composition(relative taxonomic abundances)and functional composition at KEGG module level three;(ii)physicochemical data;and(iii)antibiotics data.To preprocess compositional data,we applied a low-abundance f ilter to remove OTUs whose relative abundance did not exceed 0.01%in any sample and then log transformed the relative abundances.Likewise,for physicochemical and antibiotics data,the values of each variable were transformed to z-scores.Based on the Euclidean distances,we computed Mantel's correlations between the physicochemical data and compositional data and then the antibiotics data and compositional data(9999 permutations).We obtained the results in R(version 3.3.1)and visualized it in the Adobe Illustrator(version 16.0.0).Taxonomical composition and functional composition data were correlated to each antibiotic and physicochemical property by Mantel's tests.The distance correlations and the statistical signif icance of Mantel's r statistic corresponded to edge width and edge color,respectively[60].
Biomarker analysis
Based on their location,all water and sediment samples can be divided into two groups—impacted and less-impacted(control)groups.It is well known that the taxonomical composition of a microbial community can be impacted by local environmental variables.As a result,some bacteria might be enriched by distinctive environmental states.Linear discriminate analysis(LDA)effect size(LEf Se)[62]was used to select biomarkers in impacted and less-impacted(control)groups in water and sediment samples.Brief ly,the taxa abundance table was imported into the LEf Se pipeline,and the parameters were set as follows:the alpha value for thefactorial Kruskal-Wallis test[63]among classes and the P value for the pairwise Wilcoxon test between subclasses were both chosen to be 0.05.As to water and sediment samples,we set 3.0 and 3.5 as the threshold for the logarithmic LDA score for discriminative features,respectively.
Co-occurrence network analysis
To reduce sparsity,weselected water and sediment OTUs that werepresent in at least 50%of all water and sediment samples,respectively.We then generated separate networks for water and sediment microbial communities.The co-occurrence network was constructed using the CAVNet package(https://bitbucket.org/JackGilbertLab/cavnet)in R (as previously described by[64]).Brief ly,water and sediment networks were inferred using the Spearman correlation matrix with the WGCNA package[65].In this network,co-occurring OTUs arerepresented by nodesand connected by edges.Thenetwork deconvolution method was utilized to distinguish direct correlation dependencies[66].All P values were corrected for multiple testing using the Benjamini-Hochberg FDR-controlling procedure[67].The cutoff of the FDR-corrected P value was set at 0.01.Random matrix theory-based methods were utilized to determine the cutoff of Spearman's correlation coeff icients for water(0.84)and sediment(0.81)networks.All network properties were calculated using the igraph package in R[68].We also utilized igraph to visualize and generate water and sediment networks.The Walk Trap community detection algorithm was used to identify modules in water and sediment networks[69].To study the effect of prolonged agricultural practices,we colored each node within the water and sediment network as function of its relative abundance at impacted and less-impacted(control)sites using the‘plot_network_by_continuous_variable'function in CAVNet.
Authors’contributions
The whole study was designed by ZW and KN.MZH,JQZ,and ZW collected samples.MZH,CYC,QY,and HZ conducted DNA extraction and sequencing.MZH,CYZ,MD,and HJL analyzed the data.MZH,CYZ,MD,HJL,JG,ZW,and KN wroteand modif ied theinitial draft of themanuscript.All revised the manuscript.
Competing interests
The authors declare no competing f inancial interests.
Acknowledgments
We are grateful to Le Cao from College of Life Science and Technology,Huazhong University of Scienceand Technology,Wuhan,China,for drawing the map.This work was partially supported by the National High-tech R&D Program of China(863 Program;Grant No.2018YFC0910502),the Key Project of Hubei Province Natural Science Foundation,China(Grant No.2015CFA132),the National Natural Science Foundation of China(Grant Nos.61103167,31271410,and 31671374),and the Youth Innovation Promotion Association,Chinese Academy of Sciences,China(Grant No.2018369).
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.gpb.2018.04.008.
 Genomics,Proteomics & Bioinformatics2019年1期
Genomics,Proteomics & Bioinformatics2019年1期
- Genomics,Proteomics & Bioinformatics的其它文章
- Acknowledgments to Reviewers 2018
- GPA:A Microbial Genetic Polymorphisms Assignments Tool in Metagenomic Analysis by Bayesian Estimation
- How Microbes Shape Their Communities A Microbial Community Model Based on Functional Genes
- Inulin Can Alleviate Metabolism Disorders in ob/ob Miceby Partially Restoring Leptinrelated Pathways Mediated by Gut Microbiota
- Eあects of Proton Pump Inhibitors on the Gastrointestinal Microbiota in Gastroesophageal Ref lux Disease
- Antibiotic Treatment Drives the Diversif ication of the Human Gut Resistome