
Antibiotic Treatment Drives the Diversif ication of the Human Gut Resistome
2019-05-14 10:07:20JunLiElizabethRettedalEricvanderHelmMostafaEllabaanGianniPanagiotouMortenSommer
Jun Li,Elizabeth A.Rettedal ,Eric van der Helm,Mostafa Ellabaan ,Gianni Panagiotou *,Morten O.A.Sommer *
1 Department of Infectious Diseases and Public Health,Colleague of Veterinary Medicine and Life Sciences,City Univerity of Hong Kong,Hong Kong Special Administrative Region,China
2 School of Data Science,City Univerity of Hong Kong,Hong Kong Special Administrative Region,China
3 Novo Nordisk Foundation Center for Biosustainability,DK-2900 Hørsholm,Denmark
4 Systems Biology and Bioinformatics Unit,Leibniz Institute for Natural Product Research and Infection Biology-Hans Kno¨ll Institute,07745 Jena,Germany
5 Systems Biology and Bioinformatics Group,School of Biological Sciences,Faculty of Sciences,The University of Hong Kong,Hong Kong Special Administrative Region,China
6 Department of Microbiology,Li Ka Shing Faculty of Medicine,The University of Hong Kong,Hong Kong Special Administrative Region,China
KEYWORDS Antibiotics;Resistome;Gut microbiome;Strain;Evolution;Horizontal gene transfer
Abstract Despite the documented antibiotic-induced disruption of the gut microbiota,the impact of antibiotic intake on strain-level dynamics,evolution of resistance genes,and factors inf luencing resistance dissemination potential remains poorly understood.To address this gap we analyzed public metagenomic datasets from 24 antibiotic treated subjects and controls,combined with an in-depth prospective functional study with two subjects investigating the bacterial community dynamics based on cultivation-dependent and independent methods.We observed that shortterm antibiotic treatment shifted and diversif ied the resistome composition,increased the average copy number of antibiotic resistance genes,and altered the dominant strain genotypes in an individual-specif ic manner.More than 30%of theresistance genesunderwent strong differentiation at the single nucleotide level during antibiotic treatment.We found that the increased potential for horizontal gene transfer,dueto antibiotic administration,was~3-fold stronger in thedifferentiated resistancegenesthan thenon-differentiated ones.Thisstudy highlights how antibiotic treatment has individualized impacts on the resistome and strain level composition,and drives the adaptive evolution of the gut microbiota.
Introduction
The human intestines are densely populated by diverse microbial inhabitants,which collectively constitute the gut microbiota.About 1000 prevalent bacterial species colonize the human gastrointestinal tract,playing a pivotal role in health and disease of the host.Besides inf luencing physiology of the digestive tract,the gut microbiota also affects development,immunity,and metabolism of the host[1].External forces,including antibiotic treatment or dietary intake,shape the composition of the gut microbiota with the potential for rapid changes,thereby affecting the microbe-host homeostasis[2,3].
Antibiotics have been widely used since the Second World War resulting in dramatic benef its to public health[4].However,the rapid increase in antibiotic resistance(AR)has become an escalating worldwide issue[5].Antibiotic-resistant pathogens lead to treatment failure and contribute to increasing morbidity,mortality,and healthcare costs.Over 70%of bacteria causing hospital-acquired infections have antibiotic resistance toward at least onecommon antibiotic for treatment[6].Previous metagenomic studies have revealed the inf luence of antibiotic administration on the gut microbiota in various ways,including(1)altering the global taxonomic and functional composition or the diversity of the gut microbiota[7-11],(2)increasing the abundance of bacteria resistant to the administered antibiotic[12],(3)expanding the reservoir of resistance genes(resistome)[13],or(4)increasing the load of particular antibiotic resistance genes(ARGs)[11,14].The disruptive effects of antibiotic treatment on gut microbiota can be transient or long-lasting[15].Nevertheless,there is a paucity of information about how the resistome structure shifts and how the genotype of ARGs evolve and differentiate when the microbiome is challenged with antibiotics.In addition,how antibiotic exposure inf luences strain-level variation within the gut microbiome remains poorly understood.
Antibiotic resistance can be acquired through point mutations(de novo evolution)or horizontal gene transfer(HGT)[16].De novo resistance mutations can modify the antibiotic cellular targets or alter the expression of antibiotic resistance genes and therefore alter the resistance levels of the bacterial strain harboring them[17].Unlike de novo resistance mutations,horizontal gene transfer allows bacteria to adapt more rapidly to an environment containing antibiotics[16].Furthermore,the gastrointestinal tract,densely populated with bacteria,enables the gut microbiota to act as a resistance reservoir which likely contributes to the spread of ARGs to opportunistic pathogenic bacteria[18-20].The exchange of ARGs has been documented to occur in the human gut between strains carrying vancomycin and sulfonamide resistance genes in Enterococcus faecium and Escherichia coli,respectively[21,22].Recently,the in situ HGT of ARGs in the infant gut was described[23].However,factors triggering the HGT of ARGs in the human gut remains insuff iciently explored.Therefore,system level investigations are needed to determine whether antibiotic administration alters the dissemination potential of ARGs,and more importantly,which factors are associated with thealtered dissemination potential of ARGsin thehuman gut.
In this study,we f irst analyzed public metagenomic data from a longitudinal study of 18 cefprozil treated and 6 control volunteers[12].Wesurveyed the strain-level dynamics,shift of theresistomestructure,evolution of resistance genesat thesingle nucleotide level,and factors associated with the variation of dissemination potential.In thisf irst stageof in silico analysis wedemonstrated the diversif ication of the resistome and in situ evolution of strainsupon antibiotic intake.Wethen performed an in-depth prospective and functional study on longitudinal samples from one cefuroxime treated and one control subject using both culture-dependent and culture-independent approaches.
Results
Antibiotic treatment diversif ies the resistome composition and increases the copy number of ARGs at the intraspecies level
We analyzed public metagenomic data from a longitudinal study of 18 cefprozil treated and 6 healthy control volunteers[12](each subject was sampled at day 0,7,and 90)that aimed to investigate whether the initial taxonomic composition of the gut microbiota is associated with the reshaped post-antibiotic microbiota.Using these data we investigated the dynamics and diversif ication of the resistome,strain-level selection,variation of the dissemination potential of antibiotic resistance,and the single-nucleotide level differentiation under antibiotic treatment.
To evaluateto what extent thecomposition of theresistome was altered in response to antibiotic treatment,we quantif ied the compositional distance between samples at different time points.An NMDS plot based on the Jaccard distance of ARGs presence/absence prof ile of each sample revealed that the resistome composition was more drastically altered in the antibiotic treated group than the control group during treatment(Figure 1A).The compositional distances between baseline and treatment samples within the same treated individual were signif icantly larger than those in the control group(0.14 vs.0.06 on average,P<0.01 with Wilcoxon rank-sum test,Figure 1A).Additionally,we observed that the compositional differences between post-treatment(90 days)and the baseline samples within the same individual were also signif icantly larger in the treated group than in the control(0.135 vs.0.075 on average,P=0.04,Wilcoxon rank-sum test,Figure 1A),revealing the persistent diversif ied ARG composition after antibiotic perturbation.
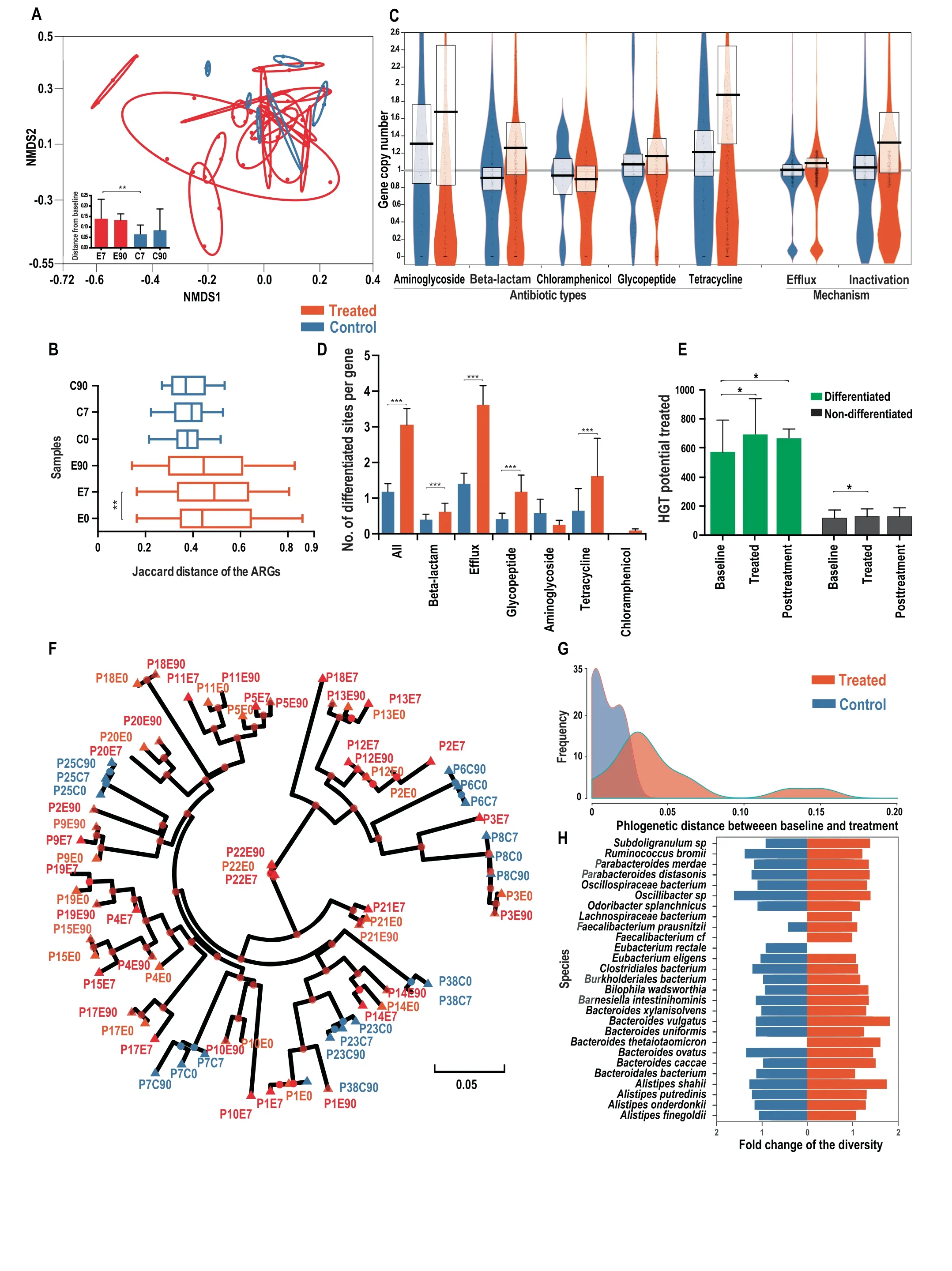
We next studied whether antibiotic treatment could select similar sets of antibiotic resistance genes and converge the resistome composition across individuals. If the gut resistome presents a more common response,instead of an individualized response,to an antibiotic treatment,we would expect the resistome compositions to become more similar after the treatment across individuals.We found that the dissimilarity of the ARGs composition measured by the Jaccard distance between individuals increased signif icantly over time in the antibiotic treated group(baseline:0.44 vs.treatment:0.52,on average,P<0.01 with Wilcoxon rank-sum test,Figure 1B),indicating that the overall composition of the resistome diversif ied under antibiotic treatment across individuals.No signif icant increase in resistome divergence was observed in the control group(P>0.05 with Wilcoxon rank-sum test,Figure 1B).When considering the ARGs abundance rather than the presence/absence,we observed a similar pattern.The Bray-Curtis distance based on the ARG abundanceprof ilebetween individualsincreased signif icantly during treatment(0.615 vs.0.685,P<0.01 with Wilcoxon rank-sum test,Figure S1).The diversif ied resistome suggests that antibiotic exposure drives an individualized selection of antibiotic resistance genes.Additional statistical analyses revealed that both the baseline species-level taxonomic composition and the baseline resistome composition are signif icantly correlated with the resistome composition after treatment(P<0.001,Mantel's test with permutation=5000).
The signif icantly diversif ied resistome composition during antibiotic treatment encouraged us to investigate whether the copy number of the ARGs(the ratio of gene depth to the relative genomic abundance of the species harboring this gene)could be altered during treatment.Incorporating the reference genomes of the 28 most prevalent species(at least 1%of relative abundance in at least half of the samples from 24 individuals,Table S1),we quantif ied the average copy number of each ARG.We found that genes annotated to confer resistance toward most classes of antibiotics,including aminoglycosides,beta-lactams,tetracyclines,and glycopeptides,increased the copy number signif icantly by 22%on average during the treatment(P<0.001,Wilcoxon ranksum test,Figure 1C).The copy number of eff lux pumps annotated as ARGs also increased signif icantly by 8%(P<0.001,Wilcoxon rank-sum test,Figure 1C).In contrast,ARGs in the control group had no signif icant increase in copy number(P=0.76,Wilcoxon rank-sum test,Figure 1C)during the same period.The signif icant increase in copy number of antibiotic resistance genes in response to antibiotic treatment could be explained by either the selection of strains with existing ARGs(one or multiple copies)or the horizontal transfer of ARGs.
Antibiotic treatment drives single nucleotide level differentiation at non-synonymous sitesNext,we investigated how the genotype of ARGs varied at the single nucleotide level within each species'population during antibiotic treatment.The analysis of single nucleotide variants(SNV)based on the shotgun metagenomic data of 72 samples from 24 individuals revealed that there is a signif icantly higher proportion of differentiated sites(see Methods for def inition)in the treated subjects than the control group(3.0%vs.1.1%,P<0.001,Wilcoxon signed-rank test,Figure 1D).There is also a signif icantly higher proportion(31%vs.13%,P<0.001 with Wilcoxon signed-rank test,Figure S2A)of genescontaining differentiated sitesin thetreated subjectsthan the controls,consistent with antibiotic exposure driving compositional changes in ARG allele frequency and spectrum.By examining sub-categories of ARGs,we observed that most genes,including those annotated as eff lux pumps or conferring resistance toward beta-lactams,tetracyclines,and glycopeptides,have a signif icantly higher proportion of differentiated sites in the treated group compared to the control(P<0.001,Wilcoxon signed-rank test,Figure 1D),indicating antibiotic-driven differentiation of the human gut resistome.
To evaluate the potential functional inf luence of the differentiated sites in the treated subjects,we investigated the frequency of differentiation at either non-synonymous sites(0-fold degenerate sites)or synonymous sites(4-fold degenerate sites).Compared with all the sites in the coding region,single nucleotide differentiation is drastically enriched at nonsynonymous sites(91.1%vs.64.9%for differentiated sites and all sites,respectively),suggesting that intraspecies level selection tended to inf luence the functional potential of the ARGs within the population of each bacterial species.
We further investigated whether there are some commonly differentiated sites in the ARGs among antibiotic treated individuals.We found that 11 genes harbored at least one recurrent differentiated site,and these genes were observed in at least 25%of thetreated individuals.Thesecommonly differentiated genes,which belong to the MexE or RND eff lux family,originate mainly from the species Bacteroides uniformis and
Bacteroides vulgatus,or other Bacteroides species(Table S2).
Figure 1 Divergence of the resistome and dominant strains in the prevalent species upon antibiotic intake
A.NMDS based on the Jaccard distance of antibiotic resistance gene prof iles(presence/absence)(stress value<0.05).E7,E90,C7,C90 represent thesamplesfrom Day 7 treatment,Day 90 post-treatment,Day 7 control,Day 90 control samples,respectively.Thered and blue ovals surround samples from the same antibiotic treated and control subjects,respectively.The radii of the ovals ref lect the compositional distancesbetween samplesfrom thesameindividual.A larger radiussuggestsa moredrastically diverged resistomeprof ile.B.Distribution of the Jaccard distance of ARGs prof ile between individuals at the same time point.C.Fold-change of the ARG copy number of major categories of ARGs.The boxes describe the mean and the standard deviation.D.Distribution of the percentage of differentiated site in various ARG categories.E.Variation of the HGT potential over time for differentiated and non-differentiated ARGs,in treated subjects at baseline,treatment,and post-treatment timepoints.F.Phylogenomic tree based on the concatenated dominant strainssequencesof 28 prevalent species.Green,red,and bluecolorsdenotethebaselineof thetreated,other timepointsof the treated,and control samples,respectively.The samples were named using the rules of‘‘P+individual ID+E/C(treated or control group)+time point(day 0,7,and 90)”,e.g.,‘‘P3E7”refers to the sample from day 7(under antibiotic treatment)from individual 3.The green color denotes the baseline of treated subject.G.Density distribution of the phylogenetic distance between baseline and treated samples in Figure 1F.A larger phylogenetic distance reveals a faster evolution under antibiotic pressure.H.Distribution of the foldchange of the genome-wide nucleotide diversity in 28 prevalent species during the treatment.*P<0.05;**P<0.01;***P<0.001.
Increased HGT potential of theresistomeisassociated with SNV differentiation
We further investigated whether the dissemination potential of ARGs was altered during antibiotic treatment.The chance of HGT transfer(HGT potential)for a particular gene is associated with two factors,the intrinsic HGT tendency of the gene and its prevalence in the population.Therefore,we estimated the HGT potential for the ARG families by combining the relative abundance and the HGT rate[24](HGT potential=HGT rate×abundance,see Methods).According to the above def inition,the gene-level variation of HGT potential is noticeably correlated with the abundance variation.We found that the HGT potential increased in both differentiated and non-differentiated ARG families during antibiotic treatment(P<0.05 with Wilcoxon signed-rank test),while the HGT potential in differentiated ARGs increased more drastically and signif icantly than in the non-differentiated ones(19%vs.5%on average,P<0.01,Wilcoxon signed-rank test,Figure 1E).This suggested that ARGs with differentiated sites could play more important roles in dissemination of antibiotic resistance upon antibiotic intake.The pattern of the differentiation-associated increase in HGT potential was not observed in the control subject at the same time period(Figure S2B).
We discovered a signif icantly positive correlation between community-level fold-change of the HGT potential and the average rate of differentiated sites in the ARGs(Pearson's correlation coeff icient 0.50,P=0.038,Figure S3),suggesting that ARGs with a higher proportion of differentiated sites tend to increase their HGT potential more drastically.One explanation for this differentiation or SNV-associated increase in the HGT potential is that ARGs with multiple genotypes coexist within the population for particular species before the antibiotic treatment and these genotypes confer distinct selective advantages or link with other benef icial alleles of ARGs under antibiotic pressure.Therefore,antibiotic treatment tends to select ARGs with selectively advantageous mutations,increasing their abundance and HGT potential.In contrast,non-differentiated ARGsgenesharboring homogeneous alleles or multiple alleles with similar selective advantage under antibiotic pressure would randomly select the strains harboring them and maintain their allele frequency.
Antibiotic pressure shifts the intraspecies population structure
The diversif ied resistome composition and single nucleotide differentiation revealed the inf luence of antibiotic treatment on thehuman gut resistome.Therefore,wefurther investigated how antibiotic pressure drives changes in the intraspecies-level population structure of intestinal species regarding the dominant strain genotypes.Wereconstructed thephylogenomic tree for the concatenated genomes of the dominant strains from 28 prevalent species(see Methods).Our results reveal that the 28 dominant strains of the treated subjects present signif icantly closer phylogenetic relationships between samples from the same individual than the samples across individuals(P<0.01,permutation test,Figure 1F),indicating an individualized antibiotic selection for the dominant strains within each subject.This individual-specif ic strain selection during treatment was also observed for each individual species,e.g.,Ruminococcus bromii(Figure S4A).Furthermore,we noticed a signif icantly higher divergence(phylogenetic distance)of the dominant strains between the baseline and treated samples in the treated group than the control(0.052 vs.0.015 on average,P<0.01,Wilcoxon signed-rank test,Figure 1F-G),suggesting that antibiotic pressure drove a signif icant shift of the dominant strain genotype,therefore inf luencing the intraspecies population structure.This is consistent with the aforementioned strong differentiation trend for the ARGs.Interestingly,when we examined the post-treatment(day 90)recovery patterns by comparing the phylogenetic distances of thedominant strains between baseline treated and post-treatment samples in the treated group,we found that the dominant strains in only 39%(7 out of 18)of the individuals had a closer phylogenetic relationship with the corresponding baseline samples than the treatment samples(Figure 1F).This suggests that the shift in intraspecies population structure upon antibiotic treatment could be long-lasting.
If antibiotic treatment tends to select dominant strains or ARGs with more similar genotypes,we should observe smaller phylogenetic distances between treatment samples across individuals,as compared with the distances between the baseline samples across individuals.The results show that there are no signif icantly different phylogenetic distances between the cross-individual treatment samples and cross-individual baseline samplesof thedominant strainsor domain ARG genotype(P>0.05,Wilcoxon signed-rank test,Figures 1F and S4B),suggesting that antibiotic treatment tends to select dominant strains and ARGs with individual-specif ic genotypic signatures.
Furthermore,we found that the differences in genomewide nucleotide diversity in 28 prevalent species are not signif icant between the treated and control groups(average fold-change:1.12 vs.1.05,P=0.73,Wilcoxon signed-rank test,Figure 1H),indicating no large-scale selective sweep or strain domination in most of the species.Interestingly,we did f ind in particular individuals and species(e.g.,Bacteroides uniformis in individuals 10,13,17,and 19)a drastically decreased(less than half of the baseline)genome-wide nucleotide diversity(Table S3),suggesting that incomplete selective sweep occured due to antibiotic exposure.Combined with the strong divergence of the dominant strains in the treated group,our analysis indicates that cefprozil intake reshapes the intraspecies population structure by selecting individualspecif ic strains and ARGs.
Cefuroximetreatment increasesantibiotic resistancelevelsof the human gut microbiota
The analyses based on public data revealed the general tendency for differentiation and diversif ication of the resistome upon antibiotic treatment.Subsequently,we prospectively and functionally assessed the impact of antibiotic treatment on the resistance levels of the gut microbiota to a panel of antibiotics in one treated and control subject using both cultivation-based multiplex phenotyping and cultivationindependent functional and shotgun metagenomics(Figure 2A and Methods).We def ined the level of phenotypically resistant gut microbiota as the ratio of colony counts on the antibioticcontaining plates to the antibiotic-free plates.The overall level of phenotypically resistant gut microbiota increased signif icantly by 140%(P<0.05,Wilcoxon rank-sum test)on theβ-lactam plates during cefuroxime treatment(day 5)(Figure 2B-C,Table S4).Even though the phenotypic resistance levels to variousβ-lactams partially recovered after the treatment,resistance never returned to their initial levels,even 3 months after treatment.In addition,the resistance levels of non-β-lactam plates showed a moderate(49%on average)but statistically signif icant increase(P<0.05,Wilcoxon rank-sum test)during treatment in the cefuroxime-treated subject(Figure 2B-C),suggesting that certain bacteria,possibly harboring multiple types of antibiotic resistance genes or multiple-antibiotic resistance genes,were selected during cefuroxime treatment.In contrast,no statistically signif icant increase in resistance was observed in eitherβ-lactam or non-β-lactam antibiotic plates in the control subject(Figure 2C).
To evaluate the variation in taxonomic composition of cultured resistant bacteria in both cefuroxime-treated and control subjects at different time points,we performed 16S rRNA sequencing of the colonies grown on different antibiotic containing plates.We observed that the community dissimilarity(beta-diversity),measured by the Morisita-Horn index,between antibiotic plates from the treated subject decreased after the cefuroxime treatment.This converging pattern of resistant communities among different antibiotic plates was not observed in the control subject(Figures 2D and S5).In addition,the pattern of converged taxonomic composition of the resistant bacteria is signif icantly stronger(P<0.01,Wilcoxon rank-sum test)onβ-lactam plates than on the non-βlactam plates(Figures 2D and S6).An NMDSplot based on the 16S rRNA taxonomic prof iles of each cultured antibiotic plate revealed that the taxonomy prof iles onβ-lactam plates weresignif icantly altered(P<0.01,permutational MANOVA test)over time(Figure2E).Such strong differentiation wasnot observed on the non-β-lactam plates(P>0.05,permutational MANOVA test).
By comparing the relative resistance of each species(16S based taxonomy)onβ-lactam plates between baseline(day 0)and treatment(day 5)samples,we identif ied 12 species with signif icantly increased phenotypic resistance during treatment(BH adjusted P<0.05 using permutation test)from all β-lactam plates in the cefuroxime-treated subject(Figure 2F).Most of these species,mainly from the genus Bacteroides,sustained enhanced resistancefor oneto threemonths(Figure2F).We further implemented the same statistical tests on nonβ-lactam plates from the cefuroxime-treated subject,as well as on bothβ-lactam and non-β-lactam plates from the control subject.No species with signif icantly increased resistance were identif ied,suggesting that the cefuroxime treatment might select for species with higher resistance to multipleβ-lactams.
Antibiotictreatmentreducesgenome-widenucleotidediversityin Escherichiacoliandenterococcusfaecium
We next investigated the temporal variation of genome-wide nucleotide diversity(clonal diversity)in different species with metagenomic analysis of the cefuroxime treated and control subjects(Figure 2A and Methods).We found that Escherichia coli and Enterococcus faecium showed exceptional variation patterns of whole genome diversity compared with all other species during cefuroxime treatment(Figure 3A).We observed strong evidence for the occurrence of genome-wide selective sweeps in these two species with a sharp decrease in genome level nucleotide diversity during treatment(Figure 3A-B),indicating that particular strains with low or intermediate initial frequency were strongly selected in the face of antibiotic pressure.These strains dominated the population with clonal expansion during antibiotic treatment and reduced the heterogeneity of the population.In addition,the temporal variation patterns between the E.coli and E.faecium werequitedifferent in terms of their post-treatment recovery mode.The genomewide nucleotide diversity of E.coli recovered gradually but
We next investigated the temporal variation of genome-wide nucleotide diversity(clonal diversity)in different species with metagenomic analysis of the cefuroxime treated and controlsubjects(Figure 2A and Methods).We found that Escherichia coli and Enterococcus faecium showed exceptional variation patterns of whole genome diversity compared with all other species during cefuroxime treatment(Figure 3A).We observed strong evidence for the occurrence of genome-wide selective sweeps in these two species with a sharp decrease in genome level nucleotide diversity during treatment(Figure 3A-B),indicating that particular strains with low or intermediate ini-tial frequency were strongly selected in the face of antibiotic pressure.These strains dominated the population with clonal expansion during antibiotic treatment and reduced the hetero-geneity of the population.In addition,the temporal variation patterns between the E.coli and E.faecium werequitedifferent in terms of their post-treatment recovery mode.The genome-wide nucleotide diversity of E.coli recovered gradually but never returned to its initial level after thecefuroximetreatment(Figure 3A),suggesting the persistence of antibiotic selected strains of E.coli.This incomplete recovery of the nucleotide diversity indicates that the f itnessof thestrain surviving antibiotic treatment was still competitive within the population in the antibiotic-free environment.In contrast,the diversity of E.faecium recovered rapidly after antibiotic treatment,suggesting that the antibiotic selected strain had a f itness cost in absence of antibiotic treatment,as indicated in previous studies[25,26].No genome-wide reduction in nucleotide diversity was observed in other species during treatment in the treated subject(Figure 3A).We observed random temporal variations of the genome-wide diversity in the control subject,which remained relatively stable over time(Figure S7),indicating that the overall strain level dynamics in the gut microbiome are more pronounced during antibiotic treatment,consistent with the observation using the public data.
Functionally selected ARGs experience strong selection at the single nucleotide level
We next adopted functional metagenomics to investigate the inf luence of the cefuroxime intake on the prevalence of different types of functionally validated ARGs over time.The results from all functional selections before treatment identif ied various types of ARGs,whileβ-lactamases increased drastically and dominated(87%of relative abundance)the recovered genes(Figure 4A-B)during the cefuroxime treatment. The β-lactamases decreased post-treatment but remained prevalent(37%)in the functional selections(Figure 4A).When quantifying the ARGs abundance from β-lactam and non-β-lactam platesseparately,wenoticed a very similar dominance ofβ-lactamases on theβ-lactam plates during the treatment(Figure S8A).Additionally,we detected an increase in eff lux genes on the non-β-lactam plates during the cefuroxime treatment period(Figure S8B),suggesting coselection of other resistance genes.This consistent with our results from the cultivation plates,where resistance levels were also enhanced on some non-β-lactam antibiotic plates.To validate the increased abundance ofβ-lactamases in the gut microbiota of the treated subject,we mapped the shotgun metagenomic data to the functionally validatedβ-lactamases.The resultsshowed thatβ-lactamasesincreased drastically during treatment in the cefuroxime-treated subject while there was no such trend in the control subject(Figure S9).

Figure 4 Variation of the gene abundance,differentiated sites,and HGT trend in the functional metagenomic ARGsA.Temporal variation of the gene abundance for functionally selectedβ-lactamases.B.Temporal variation of the gene abundance for functionally selected non-β-lactamase antibiotic resistance genes.C.Cumulative percentage of functional ARGs with different proportions of differentiated sites.The proportions of genes with differentiated sites before and during the treatment in both subjects are shown in thesubf igure.D.Proportion of genes with recent HGT signaturesand geneswith plasmid mediated HGT in both differentiated and non-differentiated ARGs.
Since the genome-wide selective sweep in E.coli and E.faecium revealed a strong selective force imposed by antibiotic treatment at the intraspecies level,we subsequently investigated further how the functional ARGs diversif ied within the population of each species during the cefuroxime treatment.The analysis of SNV,combining functional and shotgun metagenomics data,revealed that the 114 functionally selected ARGs contain a signif icantly higher proportion of differentiated sites in the cefuroxime-treated subject than in the control(0.037 vs.0.004,P<0.01 with Wilcoxon signed-rank test)(Figure 4C).At the same time,the proportion of functional ARGs with SNV signals of selection(genes with at least one differentiated site)is signif icantly higher in the treated subject than in thecontrol(56%vs.22%,P<0.01 with Fisher'sexact test)during or after treatment(Figure4C),consistent with our f indings based on the computationally annotated metagenomics datasets(Figure 1D).Further analysis revealed that the nucleotide diversity(π)of ARGs decreased signif icantly(P<0.01 with Wilcoxon signed-rank test)during treatment in the cefuroxime-treated subject(Figure S10),consistent with the strong diversif ication of the allele frequency at the single nucleotide level.
Differentiated functional ARGs tend to be recently horizontally transferred
To evaluate thetaxonomic origin and potential of recent HGT in functionally selected ARGs,we identif ied the last common ancestor(LCA)of highly homologous hits(identity>95%at amino acid level)of the functional ARGs against the NR database using blast(see Methods).We explicitly def ined recent HGT-related ARGs by considering the conf idence level of LCA and the taxonomic information from NR hits(see Methods).This analysis indicates that there is a higher proportion of differentiated functionally selected ARGs involved in recent HGT events than in the non-differentiated ones(57%vs.42%)(Figure 4D).To validate this trend of recent HGT in differentiated ARGs,we incorporated the public data described above[12]and observed a very similar pattern—the differentiated ARGs tend to be more recently transferred than the non-differentiated ones(Figure S11),suggesting a stronger HGT trend in the differentiated ARGs.
The higher rate of recent HGTs in differentiated ARGs motivated us to investigate further whether these ARGs were associated with plasmid-mediated bacterial conjugation.We searched for all highly homologous hits(>95%identity)in the NCBI plasmid RefSeq database using the ARGs as queries.We found that about 33%of differentiated and 11%of non-differentiated ARGs have highly similar(>95%identity at amino acid level)plasmid hits(Figure 4D),supporting the role of plasmids in harboring and circulating these ARGs.Since only plasmid hits with high identity(>95%)were considered,we can be quite conf ident that a substantial proportion(33%)of differentiated ARGs are actively transferred among species via plasmids in the recent evolutionary history.In our prospective study,we also observed that the HGT potential in differentiated ARGs increased more drastically than in the non-differentiated ones(304%vs.142%,P<0.01 with Wilcoxon signed-rank test,Figure S12)as we proposed above using the public dataset.
Discussion
In this study,we observed antibiotic-induced strain-level dynamics,resistome diversif ication,and increased resistance dissemination potential within the human gut.The integrative approach deployed in this study enabled the elucidation of complex dynamics of the resistome and gut microbial strains during antibiotic treatment.We used computational analyses of existing metagenomic datasetsto f ind evidencefor antibiotic induced diversif ication of the resistome,as well as substantial treatment induced differentiation of antibiotic resistancegenes.Wefurther elucidated widespread strain level dynamicsexacerbated by antibiotic treatment.Combining these results,we showed that the increased dissemination potential of antibiotic resistance gene is strongly associated with the frequency of the differentiated sites during antibiotic treatment.We then performed a functional survey in a prospective intervention study,enabling detailed culture-based and culture-independent characterization of the human gut resistome and resistance phenotypes.The consistent f indings from two experimental designs ref lect the generalization of our conclusions.
Previousstudies have provided evidence that certain factors could inf luencethe HGT potential in thehuman intestine.For instance,it has been shown that human intestinal epithelial cells produce proteinaceous compounds that modulate antibiotic resistance transfer via plasmid conjugation in E.coli[27].Another study using a mouse colitis model demonstrated that pathogen-driven inf lammation of the gut promoted conjugative gene transfer between Enterobacteriaceae species due to the transient bloom of the pathogenic Enterobacteriaceae[28].Our results revealed a similar but more comprehensive scenario about the increased HGT potential under antibiotic treatment,supporting the hypothesis that antibiotic pressure could drive the dissemination of the resistome[16].According to the def inition of HGT potential,the overall increase in ARG abundance could be the major reason for the overall increased HGT potential,although the increased ARG abundance may neither guarantee an increased HGT potential(see Methods),nor is it proportionate to overall increased abundance[24].More importantly,we observed that the increased HGT potential of ARGswascompounded by antibiotic selection of these genes at the single nucleotide level,highlighting the association between evolutionary plasticity and the horizontal transfer of ARGs[29].The single nucleotide level differentiation could be explained by the existence of a huge multiplicity of bacterial clones within single species in the gut before the antibiotic perturbation.Antibiotic treatment is the driving force selecting the alleles conferring higher survival advantage,leading to genotype differentiation and abundance increase.
Due to the fact that the disturbed microbiota could lead to adverse health outcomes[30],secondary infections[31],or increased risk of colorectal cancer[32],personalized medicine for a bacterial infection should in the future incorporate information of the patient's gut microbiota.The knowledge of the personalized gut microbiota sets the basis for predicting the stability or dynamics at the whole community level or individual bacterium upon perturbation[33].Although we cannot predict the clinical impact or the gut microbe dynamics due to insuff icient sample size and lack of clinical records in our study,our data suggest that antibiotic therapy leadsto personalized resistome diversif ication and individual-specif ic,strainlevel selection in the gut microbiota.The selective sweep observed in E.coli and E.faecium highlights the inf luence of antibiotic treatment on the intraspecies level dynamics.In line with this,future antibiotic treatment should be more personalized regarding thedosage,duration,and combination of drugs based on the unique strains and resistome composition in each patient to minimize the unintended disruption of the gut microbiota.Unfortunately,how the initial gut microbiota composition relates to antibiotic treatment eff icacy,side effects,long-term susceptibility to different pathogens,or diseases is poorly explored thus far.Therefore,more studies with longitudinal sampling and sequencing of the gut microbiota,evaluation of antibiotic eff icacy,and surveillance of susceptibility for infections or other diseases are needed.Such data could uncover the microbiota-dependent antibiotic eff icacy and side effects,the interaction networks between antibiotic and gut microbes,and long-term microbial dynamics,paving theway for future microbiome-based diagnosis and treatment.
Although our strain-level analyses offer novel insights into the dynamics of theresistome composition,copy number variation,and singlenucleotidelevel differentiation of ARGsupon antibiotic treatment,the scarcity of reference strain genomes for many species and theincompletenessof the ARG database as well as the non-robust annotation methods could potentially bias these quantif ications to certain extent and limit the generalization of our conclusions.Another caveat is that although metadata,including gender,age,weight,etc.,for each individual were provided in the original study of the public data,the sample size was not suff icient for further in-depth analyses regarding these potentially confounding factors.Future studies with more individuals,increased sampling density,and thedevelopment of morecomprehensive ARGsdatabases and accurate annotation methodologies would be of great value.
Materials and methods
Retrieval of public data
A total of 72 samples from 18 cefprozil treated subjects and 6 control subjects used in the study of Raymond et al.[12]were downloaded from NCBI SRA PRJEB8094.Each subject provided three longitudinal samples—baseline(day 0),treatment(day 7),and post treatment(day 90).
Experimental design
To functionally validate the f indings of our computational analysis based on public metagenomic datasets,two healthy adult human female subjects(age 25-29,diet not controlled)who had not taken any antibiotics for at least one year were selected for this study.One subject underwent a standard 5-day treatment with cefuroxime(500 mg,3 times a day)while the other subject had no treatment,serving as a control.Eight fecal samples were collected longitudinally over a period of three months corresponding to pre-treatment(Day 0),two time points during treatment(Days 2 and 5),one week posttreatment(Day 12),two weeks post-treatment(Day 19),three weeks post-treatment(Day 26),one month post-treatment(Day 33),and three months post-treatment(Day 97).All participants consented to these experiments and sample collections and downstream experiments and data processing followed ethical guidelines(Hvidovre Hospital)throughout the study.Samples were transported to an anaerobic chamber within an hour of collection.Fivegrams were separated out for culturing(only on Day 0,Day 5,Day 12,Day 19,Day 33,and Day 97)and 2.5 g wereused for DNA extractions.Theremaining stool samples were stored at-80°C.
Cultivation,DNA extraction,and 16S gene sequencing
The stool samples were cultivated with or without the presence of 16 antibiotics,followed by DNA extraction and sequencing of 16SrRNA as described previously[34].Brief ly,f ive grams of fecal sample was resuspended in 50 mL of prereduced(resazurin,0.1 mg/mL)1×PBS and 10-fold serial dilutions were plated on Gifu Anaerobic Media(GAM)agar with or without antibiotics in duplicate.The plates were incubated anaerobically at 37°C for 5 days.Bacterial colonies were then manually scraped off the surface of the agar and collected in 10 mL tubes.DNA was extracted from the collected samples using the MoBio UltraClean Microbial DNA Isolation kit.
Identif ication of the species with enhanced resistance from cultured plates
The relative resistance level for each OTU is def ined as the ratio of the relative abundance of the OTU from each antibiotic plate to the relative abundance of the same OTU in the control plateat each timepoint.To test whether certain OTUs had overall enhanced resistance over time,the resistance levels(e.g.,allβ-lactam plates)for each OTU were compared using a Wilcoxon signed-rank test[35]between the test time points(during or post treatment)and the baseline.
DNAextraction,library construction,and sequencingfor cultureindependent methods
The fecal samples for culture-independent methodologies were extracted using 2.5 g of sample with the MoBio Power Max Mega Soil DNA Isolation kit following the standard protocol.DNA from the treated subject's samples from Day 0,Day 5,Day 19,Day 26,and Day 33 was used to construct functional metagenomic libraries as modif ied from Sommer et al.[19].All DNA fragments from 1056 functional clones were sequenced using Sanger sequencing,imported into CLC Main Workbench where the cloning vector sequence was removed and reads of poor quality were discarded.Assembly of reads from all samples was attempted simultaneously in order to simplify the f inal output and identify sequences sharing the same sequence.All assembled contigs were hand checked for errors and correct alignment.A total of 197 contigs consisting of 2 or more sequences and 104 unassembled single sequences were retrieved.Open reading frames(ORFs)were annotated using ORFinder (http://www.ncbi.nlm.nih.gov/gorf/gorf.html).ORFs were annotated by comparing the protein(p FAM database,blastX,cdd database)or nucleotide(blastn,CARD database,ARDB database)sequences to known sequences in several databases with 80%identity and coverage.Sequence reads were removed from the annotation list if no match to known or suspected antibiotic resistance genes could be found.Forward and reverse reads from the same sequence that overlapped along the same resistance gene(s)were merged into a single annotation.
Deduction of HGTs and calculation of HGT rate
The protein sequences of thefunctional ARGsweremapped to the NCBINR database using blastp(-e 1E-5)f irst.The latest common ancestor(LCA)at species level based on highly homologous(identity>95%)hits for each ARG was deduced using MEGAN[36].By def inition,100%conf idence of the LCA at species level ref lects an explicit origin of species and 0% conf idence reveals a,theoretically,inf inite gene f low among species.An ARG was deduced as recent HGT related when all following criteria were satisf ied,(1)more than 2 highly homologous hits(identity>95%);(2)conf idence of LCA at species level less than 50%;and(3)the highly homologous hits were observed in at least two species,excluding the hits with incomplete taxonomic information at species level.To estimate the plasmid-mediated recent HGTs,we mapped all the ARGs against the NCBI plasmid RefSeq database[37]and only hits with>95%identity remained for further analysis.
A total of 154,805 gene families were retrieved from the HGTree database[38].Protein sequences of the functional ARGs were mapped to HGTree families with blastp e-value 1E-5.The HGTree family with the highest number of hits,which satisf ied the blast e-value<1E-5 and coverage>50%in the short sequenceof query and subject,was def ined as the gene family of that ARG.Phylogenetic reconciliation analysis was carried out using RANGER-DTL[39]based on the species tree and gene tree deduced by the HGTtree with optimized parameters described before[40].Only the interspecies level HGTs remained for downstream analysis.The number of HGTs was divided by the total length of the phylogenetic tree in this family to deduce the family-level HGT rate.
Shotgun metagenome library construction and sequencing
Culture-independent fecal extracts from treated subject samples Day 0,Day 2,Day 5,Day 12,Day 19,Day 26,Day 33,and Day 97 and control subject samples Day 0,Day 2,Day 19,and Day 97 were used to build shotgun metagenomic libraries using the Nextera XT kit with the standard protocol.The HiSeq 1500 was used for 100 bp PE sequencing in the CGS of The University of Hong Kong and the average throughput for each sample was 10.5 Gbp.The raw sequences can be found in BGID(CRA000815).
Quality control for the raw sequences of shotgun metagenomic data
To retrieve the high quality reads for downstream analyses,we used a series of quality control steps to remove the low quality reads/bases as described previously[41,42].In thef irst step,all the Illumina primer/adaptor/linker sequences were removed from each read.Subsequently,we mapped all the reads to the human genome with BWA version 0.7.4-r385[43],and reads with>95%identity and 90%coverage to the human genomewere removed ashuman DNA contamination.Wefurther f iltered thelow quality regions,reads,and PCR duplicates using a previously described procedure[44].
Reference mapping,gene copy number calculation,variant calling,dominant strain identif ication,and annotation of antibiotic resistance genes
The Metagenomic Intra-Species Diversity Analysis System(MIDAS)[45]was adopted to calculate the gene copy number and call singlenucleotidevariantswithin each speciesusing the default setting.To f ilter out low quality SNVs,the read error was controlled by a base quality score 30 and mapping quality were controlled by MAPQ 20 from MIDAS.The relative abundances of genes in each sample were further estimated using a RPKM measurement(number of reads per kbp length of geneper million mappablereads)using in-housescripts.The 28 prevalent species in the public shotgun metagenomic data were def ined as the species with at least 1%of relative abundance in at least half of the samples from 24 individuals(Table S1).To annotate the antibiotic resistance genes,a hidden Markov Model based prof ile searching was carried out using Resfams[46]with default parameters on the pangenome genes from MIDAS.To identify the genotypes of the dominant strain of each prevalent species at either genome level or gene level,we used the script of‘‘call_consensus.py”from MIDAS package.The phylogenetic tree was reconstructed using FastTree[47]with maximum likelihood method with default parameters.
Def inition of HGT potential
For a single gene i,the HGT potential(HGTPi)is def ined as,

where Hiis the aforementioned HGT rate in this gene family i and Aiis the relative abundance of this gene in the sample.
For a set of n genes,theoverall HGT potential isdef ined as,

According to formula(1),the increase in the HGTP for a gene is proportional to the increase in the abundance.However,for a set of genes,the overall increased abundance may not necessarily lead to an increase in overall HGTPaccording to formula(2).For example,a set of genes A,B,and C have HGT rates 0.5,1,and 2,respectively.The initial and f inal relative abundance for these three genes are(0.1,0.3),(0.2,0.3),(0.3,0.1),respectively.The overall fold-change of the abundance and HGTP is 16.7%and-23.5%,respectively.This example illustrates that the variation of the HGTP for a gene set is not only correlated to the overall abundance variation(an increased overall abundance may lead to a decreased overall HGT potential),but also thedependency between theabundance change and the HGT rate of each gene.
Detecting differentiated sites
To identify the potential adaptively evolved variant sites in the genes,we calculated the difference of the allele frequency spectrum,which was extracted from the variants calling using MIDAS mentioned above,using Fisher's exact test between treatment and baseline samples at each single nucleotide variant site.For example,the A:T ratio for a particular variant site in one ARG is 1:9 initially but was altered to 7:3 during the treatment,indicating potentially adaptive evolution where the allele frequency distribution has been differentiated.To correct for the inf luence of sequence depth on the statistical power,the maximum depth for each site was down-sampled a maximum of 10 before Fisher's exact test was carried out.The raw P values were adjusted to false discovery rate(FDR)using Benjamin's method[48].
Statistical analysis
The enhanced overall resistance level in the culture-dependent plates and the species with enriched resistance across multiple plates were analyzed using the Wilcoxon signed-rank test[35].Detection of differentiated sites according to the allele frequency spectrum and the higher proportion of differentiated sitesin the ARGsin thetreated subjectswerecarried out based on Fisher's exact test and Wilcoxon signed-rank test,respectively.The difference between resistant bacterial communities when comparing baseline and treatment plates was carried out using the permutational MANOVA test using VEGAN[49].The NMDSanalysis and the stress value calculation were performed using VEGAN.The statistical differences between groupsregarding compositional distances(Bray-Curtisdissimilarity)of gut microbiota or resistome,the Jaccard distance of resistome,the copy number of ARG families,HGT potential,phylogenetic distances,genome-wide nucleotide diversity,or nucleotide diversity of ARGs,were tested using Wilcoxon signed-rank test.The statistical correlation of the taxonomic composition and resistome prof ile was performed by Mentel's test.
Authors’contributions
MOAS,EAR,and GP designed this study.EAR performed the experiments.JL,EAR,ME,and EVDH analyzed the data.JL and EAR drafted the manuscript.All authors commented on and revised the manuscript.
Competing interests
All authors declare no conf lict of interest.
Acknowledgments
This project was supported by the Lundbeck Foundatation and EU FP7-Health Program Evotar(Grant No.282004).The study was approved(Grant No.REG-026-2014)by the Regional Ethics Committee and Danish National Medicine Agency.JL and GPwould like to thank the Centre for Genomic Sciences(CGS)of The University of Hong Kong(HKU)for their support.GP would like to thank Deutsche Forschungsgemeinschaft(DFG)CRC/Transregio 124‘Pathogenic fungi and their human host:Networks of interaction',subproject B5.We would especially like to thank Dr Agnes Chan(CGS)for fruitful discussions.We would like to thank Dr.Wendy Kwok for her language editing.
We thank David Westergaard for his involvement in the initial stageof theproject providing the ARG annotation pipeline of the shotgun metagenomics analysis.The raw sequences were deposited in BIGD(CRA000815).
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.gpb.2018.12.003.
 Genomics,Proteomics & Bioinformatics2019年1期
Genomics,Proteomics & Bioinformatics2019年1期
- Genomics,Proteomics & Bioinformatics的其它文章
- Acknowledgments to Reviewers 2018
- GPA:A Microbial Genetic Polymorphisms Assignments Tool in Metagenomic Analysis by Bayesian Estimation
- How Microbes Shape Their Communities A Microbial Community Model Based on Functional Genes
- Agricultural Risk Factors Inf luence Microbial Ecology in Honghu Lake
- Inulin Can Alleviate Metabolism Disorders in ob/ob Miceby Partially Restoring Leptinrelated Pathways Mediated by Gut Microbiota
- Eあects of Proton Pump Inhibitors on the Gastrointestinal Microbiota in Gastroesophageal Ref lux Disease