
Correlation of Gut Microbiome Between ASD Children and Mothers and Potential Biomarkers for Risk Assessment
2019-05-14 10:07:18NingLiJunjieYangJiamingZhangChengLiangYingWangBinChenChangyingZhaoJingwenWangGuangyeZhangDongmeiZhaoYiLiuLehaiZhangJunYangGuimeiLiZhongtaoGaiLeiZhangGuopingZhao
Ning Li,Junjie Yang ,Jiaming Zhang ,Cheng Liang ,Ying Wang ,Bin Chen ,Changying Zhao ,Jingwen Wang ,Guangye Zhang ,Dongmei Zhao ,Yi Liu ,Lehai Zhang ,Jun Yang ,Guimei Li*,Zhongtao Gai*,Lei Zhang *,Guoping Zhao
1 Shandong Children's Microbiome Center,Qilu Children's Hospital of Shandong University,Jinan 250022,China
2 Department of Pediatrics,Shandong Provincial Hospital Aff iliated to Shandong University,Jinan 250021,China
3 Beijing Advanced Innovation Center for Big Data-Based Precision Medicine,Beihang University,Beijing 100191,China
4 Research Institute of Pediatrics,Qilu Children's Hospital of Shandong University,Jinan 250022,China
5 Institute of Child Health Care,Qilu Children's Hospital of Shandong University,Jinan 250022,China
6 College of Life Science,Qilu Normal University,Jinan 250200,China
7 School of Information Science and Engineering,Shandong Normal University,Jinan 250358,China
8 School of Chemistry,Beihang University,Beijing 100191,China
9 CAS Key Laboratory of Synthetic Biology,Shanghai Institutes for Biological Sciences,Chinese Academy of Sciences,Shanghai 200031,China
KEYWORDS Autism spectrum disorders;Gut microbiome;Biomarker;Mother-child pair;Microbiota-gut-immunebrain axis
Abstract Variation of maternal gut microbiota may increase the risk of autism spectrum disorders(ASDs)in offspring.Animal studies have indicated that maternal gut microbiota is related to neurodevelopmental abnormalities in mouse offspring,while it is unclear whether there is a correlation between gut microbiota of ASD children and their mothers.Weexamined the relationshipsbetween gut microbiome prof iles of ASD children and those of their mothers,and evaluated the clinical discriminatory power of discovered bacterial biomarkers.Gut microbiome was prof iled and evaluated by 16Sribosomal RNA gene sequencing in stool samples of 59 mother-child pairs of ASD children and 30 matched mother-child pairsof healthy children.Signif icant differenceswereobserved in the gut microbiome composition between ASD and healthy children in our Chinese cohort.Several unique bacterial biomarkers,such as Alcaligenaceae and Acinetobacter,were identif ied.Mothers of ASD children had more Proteobacteria,Alphaproteobacteria,Moraxellaceae,and Acinetobacter than mothers of healthy children.There was a clear correlation between gut microbiome prof iles of children and their mothers;however,children with ASD still had unique bacterial biomarkers,such as Alcaligenaceae,Enterobacteriaceae,and Clostridium.Candidate biomarkers discovered in this study had remarkable discriminatory power.The identif ied patterns of mother-child gut microbiome prof iles may be important for assessing risks during the early stage and planning of personalized treatment and prevention of ASD via microbiota modulation.
Introduction
Autism spectrum disorders(ASDs)are considered a heterogeneous set of neurobehavioral disorders that are characterized by social def icits,repetitive behaviors,and cognitive inf lexibility in early childhood,which present a substantial challenge to diagnosis and treatment[1].The incidence of ASD is steadily increasing in various countries,putting a heavy burden on individuals,families,and the society[2,3].The past decade has seen good progress in identifying genetic risk factors for ASD that point to specif ic mechanisms and pathways for related behavioral def icits[4,5].Genetic studies have revealed that there is a strong genetic inf luence on the development of autism[6],however,their risk effects are highly variable,and often related to factors besides autism[7].Although such understanding should be applied to clinical nursing optimization,there is substantial discrepancy between current knowledge and clinical application [8].It has been recently recognized that genetics alone does not explain the underlying cause in many cases[9,10].Although the causes of ASD are not yet known,it is generally believed that genetic,epigenetic,and environmental risk factors interact and all play roles in the development of ASD[11,12].
Epidemiological and animal-based studies have suggested that inf lammation-induced maternal immune activation,prenatal exposure to immune challenges,and maternal obesity,stress,and gastrointestinal symptoms during pregnancy play roles in perinatal neurodevelopmental brain damage and contributeto an increased risk of subsequent neuropsychiatric disorders,such as ASDs[13-17].In recent years,therehas been a lot of epidemiological and biological evidence that prenatal factors trigger a more active immune state in the mother,which is associated with the development of autism[9,13-16,18-20].Multiple animal studies have also indicated that in addition to genetic inf luences,the maternal gut microbiota may play an essential role in the occurrence of autism in offspring during gestation,and exhibit continuous correlations and long-term pathological consequences during development[9,16,21].These studies have shown that the gut-brain axis may be formed through bidirectional communication among the nervous,endocrine,and immune systems.The imbalance in the composition and quantity of intestinal microorganisms may affect both the intestinal nervous system and the central nervous system,indicating that the microbiome-intestinal-im mune-brain axis and maternal intestinal microbiome may be pathogenic risk factors for ASD[9,18-20].Epidemiological studies have further demonstrated that alterations in the composition and metabolic products of the gut microbiome may contribute to ASD pathophysiology,and several recently published studies on autistic rodents caused by prenatal insults in femaleratssupport this view[22].Intriguingly,altering thegut microbiota and providing gut commensal bacteria(microbial reconstitution)have been shown to reverse maternal factorinduced social and synaptic def icits in offspring and have benef icial effects on ASD behaviors in both mice and humans[9,16,21].Evidence for the link between ASD and abnormalities in gut microbial function hasbeen accumulating;however,epidemiological and animal studies alone may not be suff icient enough to determine the actual correlations and mechanisms in humans[18].
Multiple studies using clinical samples have reported differences in microbiota composition and specif ic enteric bacteria existing in fecal,ileal,or duodenal samples from ASD children,providing potential diagnostic and therapeutic targets[23-34].The majority of these studies focused more on evidence-based correlation analysis of microbiome with ASD than causality questions,using cohorts of mostly Caucasians.Varioustypesof gut microbeshavebeen identif ied asbiomarkers,such as Clostridium tetani,Desulfovibrio spp.,Bacteroides vulgatus,Alcaligenaceae,Sutterella spp.,Ruminococcusgnavus,Ruminococcus torques,Lactobacillus spp.,Desulfovibrio spp.,Fusobacteria, Verrucomicrobia, Eubacteriaceae, Lachnospiraceae,Sutterellaceae,Enterobacteriaceae,Bif idobacterium, Faecalibacterium, Prevotella, Coprococcus,unclassif ied Veillonellaceae,Burkholderia,Neisseria,Alistipes,Bilophila,Dialister,Parabacteroides,Veillonella,Collinsella,Corynebacterium,and Dorea.A recent study has investigated the differences between fecal microbial communities of 35 Chinese ASD children and 6 typical development(TD)children.No difference in alpha diversity was found between the two groups,while relative abundance of Sutterella,Odoribacter,and Butyricimonas in the ASD group wasmuch higher.In contrast,abundance of Veillonella and Streptococcus was signif icantly reduced compared to that of the TD group[35].However,to the best of our knowledge,no study has investigated the gut microbiome prof iles of mother-child pairs of ASD children and evaluated their correlations at the same time.Thus,it is still unclear how the gut microbiome varies between mothers of ASD children and those of healthy children and whether maternal gut bacterial communities areassociated with the gut microbiome prof iles of ASD children.Additionally,the unique features of the gut microbiome of ASD children in comparison with their mothers or healthy children have not yet been identif ied.Therefore,in this study,we examined the relationships among the gut microbiome prof iles of ASD children and their mothers and evaluated the potential clinical discriminatory power of thediscovered bacterial biomarkers.Thispilot study suggeststhe roleof gut microbiota in autism and could serve as a basis for further investigation of the combined effect of genetic,microbial,and hormonal changesfor development and clinical manifestation of autism.
Results
Study population
We enrolled 59 ASD children(ASD-Cs)and their mothers(ASD-Ms),together with 30 matched healthy(neurotypical)children(H-Cs)and their mothers(H-Ms),for the current gut microbiome study.The average age of ASD-Cs and HCs at the time of sample collection was 4(range,2-7)and 5(range,2-10)years old,respectively.The average age of ASD-Ms and H-Ms at the time of sample collection was 33(range,26-38)and 31(range,27-42)years old,respectively.The characteristics of the children and mothers are reported in Table 1 and Table S1.In the subsequent results and analyses,we def ined the groups as ASD-C,ASD-M,H-C,H-M,ASD-M+C(for mother-child pairs of ASD children),and H-M+C(for mother-child pairs of healthy children).
ASD children harbored an altered gut microbiomein the Chinese cohortFor characterization of the gut microbiome associated with ASD,we compared the alpha diversity between the ASD-C and H-C groups.We found signif icant increases in bacterial richness(P<0.01,Figure 1A and Figure S1D)in ASD-Cs and nonsignif icant difference in bacterial diversity between two groups(P=0.13,Figure S1A).To further explore the characteristics of intestinal bacterial community of ASD-Cs,we assessed the relative taxon abundance in the microbiota of the ASD-C and H-C groups.The total distribution of bacterial taxonomy showed no signif icant variations in the bacterial communities between ASD-Cs and H-Cs at the phylum level,as characterized by a similar Firmicutes/Bacteroidetes ratio(P>0.05,Figure 1D);however,a signif icant increase in the relative abundance of Proteobacteria was observed in the ASD-C group compared with the H-C group(P<0.01).We also compared differences in the taxa at the genus level(Figure S2A).Analysis of the beta diversity based on the unweighted UniFrac distances showed that the microbiome of the ASD-C group was distinct from that of the H-C group.We further performed an analysis of similarities(ANOSIM),and the results indicated that the structure of the gut microbiome of the ASD-C group was signif icantly different from that of the H-C group(ANOSIM,r=0.197,P<0.01,unweighted UniFrac,Figure 2A).
Linear discriminant effect size(LEfSe)analysis between the ASD-C and H-C groups revealed the signature microbiome prof iles and predominant bacterial biomarkers of ASD children.Signif icant increases in the relative abundance of Enhydrobacter, Chryseobacterium, Streptococcus, and Acinetobacter(at the genus level),as well as Acinetobacter rhizosphaerae and Acinetobacter johnsonii(at thespecies level),in addition to a signif icant reduction in Prevotella melaninogenica(at thespecies level),wereobserved in the ASD-C group in comparison with the H-C group,as indicated by the lineardiscriminant analysis(LDA)(LDA score>3,Figure 3A).All potential biomarkers(LDA score>2)are shown in Figures S3A and S3D and listed in Table S2.
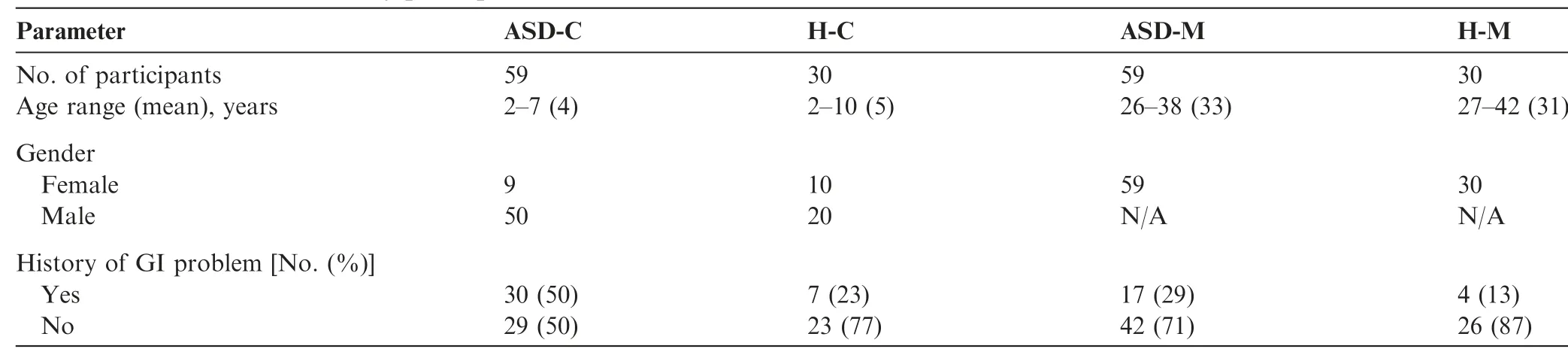
Table 1 Characteristics of study participants

Figure 1 Comparison of the alpha diversity and relative abundances at the phylum level based on the OTU prof ileComparison of the alpha diversity was evaluated using PD_whole_tree based on the OTU prof ile between the autism groups and the control groupsand shown in thetop panelsfor ASD-C vs.H-C(A),ASD-M vs.H-M(B),and ASD-M+C vs.H-M+C(C).P valueswere calculated using the Wilcoxon rank-sum test.The relative abundances of different taxa at phylum level were shown in the bottom panels for ASD-C vs.H-C(D),ASD-M vs.H-M(E),and ASD-M+C vs.H-M+C(F).OTU,operational taxonomic unit.ASD-C,ASD child;ASD-M,mother of ASD child;H-C,healthy child;H-M,mother of healthy child;ASD-M+C,mother-child pair of ASD child;H-M+C,mother-child pair of healthy child.
Mothers of ASD children harbor an altered gut microbiome
To identify the differences in gut microbiome between mothers of ASD-Cs and H-Cs,we compared the alpha and beta diversities between ASD-Ms and H-Ms.Distinct gut microbiome prof iles were revealed.Analysis of the alpha diversity showed a signif icant increase in bacterial richness of ASD-Ms(P<0.05,Figure 1B and Figure S1E)and nonsignif icant difference in bacterial diversity(P=0.35,Figure S1B).We also analyzed the relative abundance of microbes in the gut microbiome between ASD-Ms and H-Ms at the phylum level(Figure 1E)and genus level(Figure S2B).Then,the analysis of beta diversity based on the unweighted UniFrac distances revealed that the microbiome of ASD-Ms was signif icantly different from that of H-Ms(ANOSIM,r=0.248,P<0.01,unweighted UniFrac,Figure 2B).LEfSe analysis further conf irmed these signif icant differences.Notably,a signif icant increase in the relative abundances of Moraxellaceae and Enterobacteriaceae(at the family level)and Acinetobacter(at the genus level)and a signif icant reduction in Faecalibacterium were observed in the ASD-M group,in comparison with the H-M group(LDA score>3,Figure 3A).All potential biomarkers(LDA score>2)are shown in Figures S3B and S3E and are listed in Table S2.
ASD children harbored unique bacterial biomarkers
For characterization of the gut microbiota between motherchild pairs,we compared the alpha and beta diversities between ASD-M+C and H-M+C groups,and again revealed distinct gut microbiome prof iles.Analysis of alpha diversity showed that the sequence-based boxplot based on the PD_whole_tree was nearly asymptotic,and Wilcoxon ranksum tests demonstrated signif icant differences in diversity in the ASD-M+C and H-M+C groups(P<0.01,Figure 1C).Abundance-based coverage estimator(ACE)indexesalso conf irmed these f indings(Figure S1F).However,there was no signif icant difference in Shannon index(Figure S1C).Analysis of beta diversity based on the unweighted UniFrac distances revealed that the microbiome of the ASD-M+C group clustered apart from that of the H-M+C group(ANOSIM,r=0.191,P<0.01,Figure 2C).We then used the identif ied signif icant bacterial biomarkers to evaluate the correlations and revealed distinct clustering of children and mothers'gut microbiomes in the two groups(Figure 3A,Figures S3C and S3F).We further analyzed the different gut microbiome structures between ASD children and their mothers and found that relative abundance of genus Clostridium was increased in ASD children(Figure 4).
Furthermore,the number and identity of the shared operational taxonomic units(OTUs)were evaluated through Venn diagrams.As shown in Figure 2D and Table S3,the similarity of the gut microbiome in the ASD-M+C group was higher than that in the H-M+C group.Bacteroides ovatus and Abiotrophia were found in the H-M+C group.Epulopiscium,Sphingobium xenophagum, Anaeroplasma, Adlercreutzia,Solirubrobacterales,Mesorhizobium,Hydrogenophilus,Salinicoccus,and Promicromonosporaceae were only found in ASD children.
Figure 2 Microbiome community and Venn diagram analysisPCoA of bacterial beta diversity based on the unweighted UniFrac distances.ASD-C vs.H-C(A);ASD-M vs.H-M(B);ASD-M+C vs.H-M+C(C).D.Venn diagram displaying the degree of overlap of bacterial OTUs among ASD-C,ASD-M,H-C and H-M.PCoA,principal coordinate analysis.
The discovered bacterial biomarkers may have potential value in risk assessment
The relative abundance of the top 96 most different OTUs across groups was determined using the criteria of LDA score>2 and adjusted P<0.1 by Wilcoxon rank sum test(Figure 3A).The signature OTUs across groups at different P value stringency are marked in Figure 3A,and the number of these OTU signatures are summarized in Table S3.According to stringent criteria for adjusted P value(P<0.01,Wilcoxon rank sum test)and LEf Se analysis(LDA score>3),candidate biomarkers were selected to predict the risk of disease in children with autism;candidate biomarkers were also selected to distinguish between mothers of ASD children and healthy children.Five biomarkers were selected for ASD-C vs.H-C,including Betaproteobacteria,Burkholderiales,Pseudomonadales,Moraxellaceae,and Acinetobacter;six biomarkers were selected for ASD-M vs.H-M,including Flavobacteriia,Gammaproteobacteria, Flavobacteriales, Weeksellaceae,Enterobacteriaceae,and Enterobacteriales.
To explore the potential value of the identif ied bacterial biomarkers for two levels of clinical discrimination(ASD-C vs.H-C,and ASD-M vs.H-M),we constructed receiver operating characteristic(ROC)curves and computed the area under the curve(AUC)values.For ASD-C vs.H-C,the highest AUC value was 0.944(4-fold,95%conf idence interval[CI]:84%-100%)with 83.33%sensitivity and 91.67%specif icity(Figure 3B).For ASD-M vs.H-M,the highest AUC value was 0.986(4-fold and 5-fold,95%CI:94%-100%),with 100%sensitivity and 100%specif icity(Figure 3C).The evaluation of all biomarker combinations is summarized in Table S4.Additionally,we evaluated the effect of age,sex,and history of gastrointestinal(GI)problem on the f ive candidate biomarkers for ASD-C vs.H-C and the effect of age and history of GI problems on the six candidate biomarkers for ASD-M vs.H-M.None of these factors had signif icant effects on the selected candidate biomarkers(Tables S5 and S6).

Figure 3 The relative abundance of the OTUs and ROC curves A.The relative abundance of the top 96 most different OTUs across groups(LDA score>2 and adjusted P<0.1)according to the Wilcoxon rank sum test.The abundance prof iles are transformed into Z scores by subtracting the average abundance and dividing the standard deviation of all samples.Z scoreisnegative(shown in blue)when theraw abundanceislower than themean.OTUswith adjusted P<0.01 and P<0.05 are marked in red and green,respectively.B.ROC curve with adjusted P<0.01,(Wilcoxon rank sum test)and LDA score>3(LEfSe analysis).5 biomarkers were selected to predict the risk of disease in children with autism.These include Betaproteobacteria,Burkholderiales,Pseudomonadales,Moraxellaceae,and Acinetobacter.C.ROC curve with adjusted P<0.01(Wilcoxon rank sum test)and LDA score>3(LEfSe analysis).6 biomarkers were selected to predict the risk of disease in children's mothers.These biomarkers are Flavobacteriia,Gammaproteobacteria,Flavobacteriales,Weeksellaceae,Enterobacteriaceae,and Enterobacteriales.The SVM classif ier from R package e1071 was adopted to perform the classif ication analysis for the selected biomarkers.Five-fold cross-validation was used to evaluate theperformance of the predictivemodel.The ROC curvesaswell asthe AUC value was calculated using the ROCR R package.P values were adjusted by FDR.ROC,receiver operating characteristic;LDA,linear discriminant analysis;LEfSe,linear discriminant effect size;AUC,area under the curve;FDR,false discovery rate.
KEGG pathwaysweredistinct between gut microbiomeof children,mothers,and mother-child pairs
The phylogenetic investigation of communities by reconstruction of unobserved states(PICRUSt)method was used to predict the KEGG pathways between the microbiome of ASD patients and healthy subjects,and 40 different KEGG pathways were found.The ASD-C group showed increased activities in some disease pathways,such as pertussis,amyotrophic lateral sclerosis,and Parkinson's disease,and in bacterial motility proteins.There were 38 different KEGG pathways identif ied in the ASD-M and H-M groups.The ASD-M group showed increased enrichment in pathways in amyotrophic lateral sclerosis,bacterial motility proteins,renal cell carcinoma,and geraniol degradation(Figure 5).
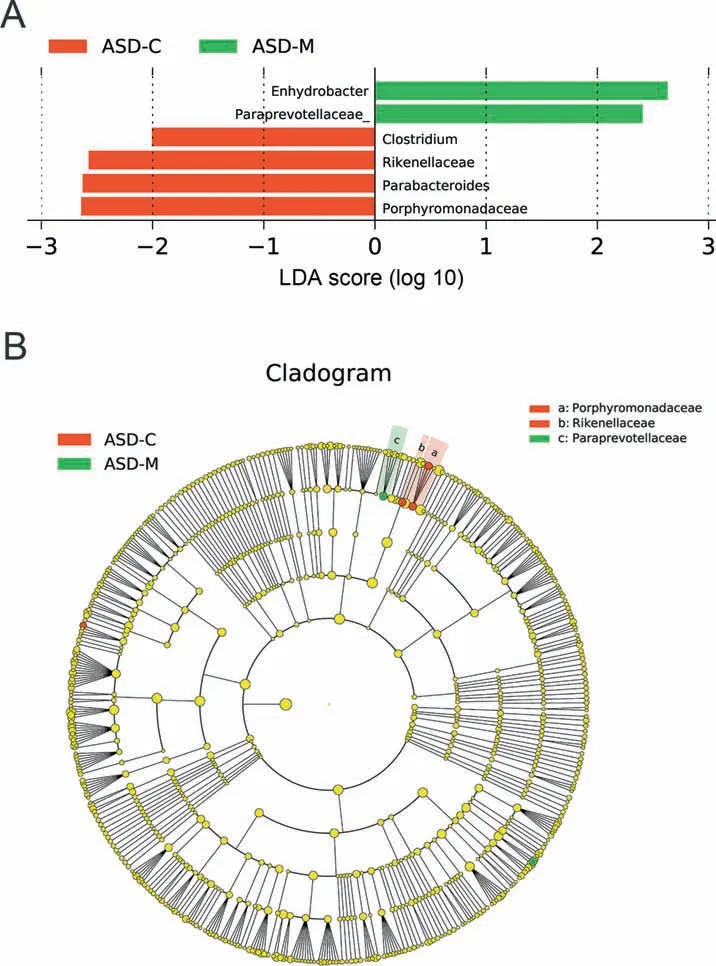
Figure 4 LEfSe analysis between ASD-C and ASD-M groupsA.Histogram of the LDA scores computed for differentially abundant taxa between ASD-C and ASD-M.The LDA score indicates the effect size and ranking of each differentially abundant taxon.B.The enriched taxa in ASD-C and ASD-M gut microbiomerepresented in the cladogram.The central point represents the root of the tree(Bacteria),and each ring represents the next lower taxonomic level(phylum to genus:p,phylum;c,class;o,order;f,family;g,genus).The diameter of each circle represents the relative abundance of the taxon.
Discussion
Many recent studies have conf irmed that the causes of autism include genetic and environmental factors[13-16].However,research into the environmental factors associated with the development of autism and the molecular mechanisms by which these factors operate is just beginning.Our current study is a pilot exploration to examine the gut bacterial diversity of ASD children using a Chinese cohort,to examine the gut bacterial diversity of the mothers of ASD children,and most importantly,to discover correlations between the microbiome prof iles of ASD children and their mothers.We found that the gut microbiota of Chinese ASD children showed signif icant change,including increased bacterial richness,and different microbiota structures,compared with healthy children.In addition,there were close correlations between the microbiome prof iles of mother-child pairs,and ASD children exhibited unique bacterial biomarkers.Finally,the identif ied bacterial biomarkers exhibited remarkable discriminatory power for differentiating ASD children from neurotypical control children,as well as their mothers.

Figure 5 Predicted metagenome function based on KEGG pathways analysis Extended error bar plot showed the signif icantly different KEGG pathways between ASD-C and H-C(A),between ASD-M and H-M(B),between ASD-M+C and H-M+C(C).
ASD children harbored an altered gut microbiome
Consistent with previous clinical studies on the gut microbiomeof ASD children,a comparison of thegut bacterial community structures between ASD children and healthy controls in our study revealed signif icantly shifted microbiome prof iles in thefeces of ASD children.Further,it identif ied a set of bacterial biomarkers that varied signif icantly between the two groups.Previous studies have suggested that the gut microbiome of autistic children may contain harmful genera or species that contribute to the severity of autism symptoms(Tables S7 and S8).A comparison of our results with thoseof previous studiesusing other ethnic cohortsfound similarities and differences,indicating that the gut microbiome prof ile and key bacterial signatures may have potential commensal causative microbes and ethnicity-specif ic characteristics.These potential commensal causative microbes identif ied in our study and previous studies belong to different taxa.A detailed comparison of the similarities and differences in biomarkers can be found in the Supplemental Material.We have compared our results with one of the most recently published articles on the gut microbiome of Chinese ASD children[35],which examined the differences in fecal microbiota in 35 children with ASD and 6 TD children.In addition to their f indings,our study moved one step forward to f ind correlations between the gut microbiome of ASD children and their mothers.Notably,Clostridium and Streptococcus were found to be increased in ASD children in our study and most previous studies,in which the potential functions of these two genera have been extensively discussed [23,28,36-38].Clostridium,Streptococcus,
Chryseobacterium,Haemophilus,and Comamonas are involved in GI disorders,maternal inf lammation,maternofetal immune activation,neonatal sepsis,bacteremia or meningitis,acute appendicitis,and childhood vaccination[38-42].Williams et al.demonstrated the presence of members of the family Alcaligenaceae in some autism children,but there were no Alcaligenaceae sequences detected in the microbiota from healthy children[29].Our study also found a signif icant increase in the abundance of Alcaligenaceae in the gut microbiota from the ASD-C group compared with that in the H-C group.Interestingly,we discovered an evidential increase in Parabacteroides johnsonii,a Gram-negative and obligate anaerobe,in the feces from autism children.The genus Parabacteroides has been reported to have increased abundance in the feces of ASD children[32].However,its connection with the pathogenesis of autism remains to be further investigated.
The distinct gut bacterial biomarkers of Chinese ASD children also included four species showing increased abundance,namely,A.johnsonii,A.rhizosphaerae,Brevundimonas diminuta,and Stenotrophomonas geniculate.If these unique microbes are found to be causative or consequential factors in Chinese patients with ASD,such f indings may facilitate thedevelopment of specif ic diagnostic tests as well as strategies for treatment and prevention of ASD.
Gut microbiome was different in the mothers of ASD children
As we hypothesized,the gut microbiome varied dramatically between the mothers of ASD children and healthy children.Because the mothers of ASD children were neurotypical,we could only attribute variations in their bacterial biomarkers,e.g.,increases in Streptococcus and Acinetobacter and decreases in Porphyromonas and P.melaninogenica,to potential GI disorders,maternal inf lammation,bacteremia,and antibiotic usage during pregnancy;however,further research is needed to determine the specif ic causes of these changes.Importantly,we discovered a striking correlation between the microbiomes of mother-child pairs.Previous studies have suggested vertical transmission of the microbiome from the mother to the gut of offspring based on their similarities[43],which could partially explain the similarities in the gut microbiome prof iles of the mother-child pairs.In contrast,ASD children harbored unique bacterial biomarkers when compared with their mothers,indicating the potential roles of these microbiota in the etiology of autism.For example,Alcaligenaceae,Clostridium,Haemophilus,and Wautersiella were increased only in the ASD-C group and not in the ASD-M group,whereas Ruminococcaceae and Paraprevotellaceae were decreased only in the ASD-C group and not in the ASD-M group.OTU comparisons also showed that ASD children had unique bacterial biomarkers,such as Epulopiscium,S.xenophagum,Anaeroplasma,Adlercreutzia,Solirubrobacterales, Mesorhizobium, Hydrogenophilus,Salinicoccus, Corynebacterium variabile, and Promicromonosporaceae.
Biomarkers may have predictive power on autismIn the current study,Bacteroidetes and Firmicutes were demonstrated to be important phyla.It is worth noting that the vast majority of species in the Bacteroidetes produce propionic acids and other short-chain fatty acids as f inal products of their metabolism.MacFabe and colleagues have shown clearly when injecting propionic acid or other short-chain fatty acids into rat cerebral ventricles,rats showed unique biological,chemical,and pathological changes,which were the characteristic of autism[44].Decreasing harmful populations with antibiotics such as vancomycin have been shown to be an important step in improving the symptomsof lateonset autism[23,45].
Signif icant increases in the abundance of species belonging to Proteobacteria were found in the microbiome of ASD group.Proteobacteria include abundant gram-negative pathogens such as Escherichia,Salmonella,Vibrio,Helicobacter,and Yersinia[46],which induce inf lammatory responses through lipopolysaccharide(LPS)on the cell wall.Previous studies revealed that lipopolysaccharides(LPS)treatment of the schwannoma in vitro improved the level of NFκB,IL-1β,pSTAT3,and IL-6 cytokines to activate an immune reaction.Additionally,the connection of Proteobacteria with chronic enteritis was demonstrated based on a mouse model[47].
Clostridium and Streptococcus were found to be increased in ASD children in our study and most previous studies,in which the potential of these two genera have been extensively discussed[23,28,36-38].Parracho et al.described the increased abundance of Clostridium in the stool from autistic patients based on f luorescent in situ hybridization analysis[33].A possiblemechanism of autism pathogenesis isthat neurotoxin produced by several Clostridium bacteria transits through the vagus nerve into the brain and then blocks neurotransmitter delivery to cause children's abnormal behavior.In addition,the evident increase in the abundance of pathogenic gener a Wautersiella,Agrobacterium,Chryseobacterium,Streptococcus,and Acinetobacter was found in the gut microbiome of the ASD group.Wautersiella has been isolated from various samples,including wound samples,blood samples,respiratory samples from patients with cystic f ibrosis,samples from suspected joint prosthesis infection,and the urine of pyelonephritis infants[48].Agrobacterium species are able to infect immunocompromised children to cause bacteremia[49].Chryseobacterium species infect immunocompromised neonates and adults to cause neonatal sepsis,bacteremia,or meningitis,which can be associated with autism pathogenesis[40].The connection of Streptococcus with neurological disorders described in previous studies is consistent with the improved abundance in the ASD group in the current study.Acinetobacter spp.leads to serious infections,including sepsis,pneumonia,meningitis,endocarditis,skin infection,and wound infection[50-53].
We speculated that the decreasing abundance of Prevotella[54]and Ruminococcus could cause autism pathogenesis through blocking arginine(Arg)metabolism.Argininemia caused by excessive Arg in the blood may lead to neurodegeneration.Further,considering that Arg is one of the substrates of citrulline synthesis,the increase in nitric oxide produced by high levels of Arg-activated citrulline synthesis may inhibit proliferation and differentiation of neural stem cells as neurotoxins[55,56].Interestingly,the onset time(the third month to the fourth year)and symptom(the loss of cognitive and athletic competence)of argininemia have striking similarity with autism[57,58].In addition,the species of Prevotella and Ruminococcus were found to be involved in Arg metabolism[59]and had decreased abundance in the microbiome of ASD group in our study.These facts indicated that the suppressed metabolism of Arg caused by reducing Prevotella[54]and Ruminococcus could lead to high-level nitric oxide,which might cause abnormal neural development and the onset of autism.
Limitation
Screening for autism carries two major challenges.The f irst is the need to predict or detect autism in early childhood,even before the onset of symptoms.The second is the ability to differentiate pregnant women at high risk of having autistic children.Based on the discovered biomarkers,ASD children and their mothers could be separated from healthy children and their mothers with high sensitivity and specif icity.Thediscriminatory power of these candidatebiomarkerspavesthe way for establishing fecal microbiome tests for clinical diagnostic and prognostic screening for ASD.However,our study has some limitations.First,the cross-sectional nature of the study prevented us from elucidating the mechanisms and longitudinal view of relevance.Additional large cohort studies are needed to determine the chronological order and to evaluate changes in the gut microbiota of mothers and children.In addition,small sample sizes did not allow subgroup analysis to assess whether the associations of different ASD patients are consistent,as def ined by factors such as severity and comorbidities.Separate test and validation cohorts are needed in order to draw the useful conclusions about the discriminatory power of the microbial biomarkers.However,the potential value of these biomarkers for clinical validation and application is not diminished,and nested case-control studies using population-based cohorts are currently under way.
Conclusions
We found signif icant differences in the composition of intestinal bacteria between ASD children and healthy children.Although the gut microbiome of ASD children was closely associated with that of their mothers,children with ASD still had unique bacterial biomarkers.Variation of maternal gut microbiota may play a critical role in increasing the risk of ASD in children.The identif ied similarities and differences in mother-child gut microbiome prof iles are important for early assessment of risks and for planning personalized treatment and prevention strategies for ASD via microbiota modulation.Our current study has several noteworthy weaknesses.There was no assessment of diet quality,especially f iber intake effects,on the gut microbiome.The factorsof living conditions and history of GI problems were not properly controlled and fully evaluated.Most importantly,a longitudinal study and large cohort validation are warranted to monitor the variation of the gut microbiome of ASD children and their mothers.
Materials and methods
Study participants
In this study,which was approved by the Institutional Review Boards of Qilu Children's Hospital of Shandong University(QCH IRB#16-015)and Shandong Provincial Hospital Aff iliated to Shandong University(SPH IRB#16-0061),sample collection began in July 2016.All participants(mothers)who visited the Institute of Child Health Care and agreed to serve as fecal donors provided written informed consent and questionnaire data sheets,in accordance with national legislation and the Code of Ethical Principles for Medical Research Involving Human Subjects of the World Medical Association(Declaration of Helsinki).The specimen bank for the Children's Microbiome Initiative at Qilu Children's Hospital,in collaboration with the Shandong Provincial Hospital,has collected over 2000 samples from individuals with various diseases and healthy individuals. Parental samples were obtained whenever possible.From this specimen bank,we used stool samples from 59 mother-child pairs of ASD children and 30 matched mother-child pairs of healthy(neurotypical)children for the current gut microbiome study.
ASD patientswith clinically signif icant inf lammatory symptoms were excluded.We also collected the clinical index of amino acid level,including alanine,glycine,proline,leucine+isoleucine,valine,methionine,phenylalanine,tyrosine,citrulline,ornithine,Arg(Figure S5).Patients with ASD were consecutively admitted to the Instituteof Child Health Careof the Qilu Children's Hospital,and ASDs were diagnosed according to the Diagnostic and Statistical Manual of Mental Disorders,5th Edition(DSM-5)[60],and evaluated using the Autism Diagnostic Observation Schedule and Autism Behaviour Checklist and the proposed criteria for ASD in the DSM-5 [61].All participants in this study received a Chinese-based diet provided daily by the hospital cafeteria,and no antibiotics,probiotics,or prebiotics had been taken within 3 months before sampling.No patients were treated with anti-inf lammatory or antioxidant drugs.The Institute of Child Health Care of Qilu Children's Hospital is a center specialized for training and treatment of ASD.ASD children included in the current study were all from the same sixmonth training class,during which they stayed in the hospital ward,atethehospital meal provided daily by thehospital cafeteria,and received training together with their mothers.At the same time,the Research Institute of Pediatrics of Qilu Children's Hospital conducts another clinical study,for which healthy children and their parents(hospital employees including doctorsand nurses)were surveyed by having thesamemeal provided by the hospital cafeteria for three months.All stools were sampled at the end of the third month.
Sample collection,DNA extraction,and sequencing
Stool samples from enrolled patients were collected with sterilized 2-ml tubes containing pure ethanol,aliquoted,and frozen at-80°C until DNA extraction.Total DNA extraction from fecal samples(250 mg,wet weight)was performed using a FastDNA SPIN Kit for Feces(MPBiomedicals,Santa Ana,CA,USA)according to the manufacturer's instructions.NanoDrop ND-1000 Spectrophotometer(Nucliber)was used for DNA quantif ication with an equivalent of 1μL of each sample.For each DNA sample,we amplif ied respectively the bacterial 16S rRNA genes using a primer set specif ic for V1-V2 variable region of 16S rRNA gene with the universal primers F27(5′-AGAGTTTGATCMTGGCTCAG-3′)and R338-I(5′-GCWGCCTCCCGTAGGAGT-3′)and R338-II(5′-GCWGCCACCCGTAGGTGT-3′).Amplicons were f irst purif ied using the QIAquick PCR Purif ication Kit(Qiagen,Barcelona,Spain),quantif ied using a NanoDrop ND-1000 Spectrophotometer(Nucliber)and then mixed at the same concentration.The mixed amplicons(2 nM)were then sequenced by Illumina HiSeq sequencer(Illumina Inc.,San Diego,CA,USA),as described in the standard Illumina platform protocols.In this study,all sequencing data were uploaded to the NCBI SRA database(accession number:PRJNA453894)and can be accessed at https://www.ncbi.nlm.nih.gov/sra/.All sequencing data can also be viewed at NODE (http://www.biosino.org/node)by pasting the accession No.OEP000294 into the text search box or through the URL: http://www.biosino.org/node/project/detail/OEP000294.
Analysis of 16S rRNA gene sequences
Raw FASTQ f iles were processed demultiplexed,qualityf iltered by Trimmomatic according to the previous description[62].The f iles were then merged by FLASH with the following criteria.(a)When an average quality score<20 was obtained on a 50 bp sliding window,all readers were truncated at any site.(b)Primers were exactly matched and allowed for a 2-nucleotide mismatch.Deleted readers contained ambiguous bases.(c)Merged sequences that are longer than 10 bp in overlap based on overlapping sequences. High-throughput sequencing analysis of bacterial rRNA genes was processed using the Quantitative Insights into Microbial Ecology(QIIME,version 1.9.1)software suite[63],according to the QIIME tutorial(http://qiime.org/).Chimeric sequences were,subsequently,removed using usearch61[64]with de novo models.Selected high-quality sequences were clustered against the 2013 Greengenes(13_8 release)ribosomal database(97%reference data set).Sequences that did not hit the reference sequence collection were subsequently assigned to de novo OTUs with UCLUST algorithm in QIIME with a similarity threshold of 97%.The taxonomic identity of each OTU was determined using the RDP Classif ier[65]within QIIME and the Greengenes reference data set.Alpha and beta diversity metrics from the f inal OTU table without singletons were obtained within the QIIME pipeline.Principal component analysis(PCA)was complemented by hierarchical clustering using unweighted pair group method with arithmetic mean(UPGMA)clustering(also known as average linkage)on the distance matrix of OTU abundance.The QIIME package was used to obtain a Newick formatted tree.
The LEfSewasused to explorepotential bacterial biomarkers associated with different groups.LEf Se is an algorithm for high-dimensional biomarker discovery,which uses LDA to estimate the effect size of each classif ication unit that differs between cases and controls.Besides detecting signif icant features,LEfSe also ranks features based on effect size,putting features that account for most of the biological difference at the top[66].The selected biomarkers were classif ied and analyzed by SVM classif ier of R package e1071.The performance of the predictive model was evaluated using f ive-fold crossvalidation.The ROCR R package was used to calculate the ROC curve and the AUC value[67].
Statistical analysis
In order to account for any bias caused by nonuniformity sequencing depth,the minimum number of sequences present in any given sample from a sample category was selected randomly before calculating community-wide dissimilarity measures(alpha diversity and beta diversity),and we raref ied the sequence data in QIIME to a sequencing depth of 11,000 per sample for both diversity analyses.Principal coordinates were computed for the unweighted distance matrices and used to generate principal coordinate analysis(PCoA)plots using evenly sampled OTU abundances.Based on the marker gene data and a database of reference genomes,the functional composition of a metagenome was predicted with PICRUSt,as described by Langille et al.[68].Graphical representations of the results were created using STAMP[69]and the calculation of P values was performed with Kruskal-Wallis H-tests and Welch's t-tests.The P values were corrected by False Discovery Rates(FDR)to control for multiple hypothesis testing.Benjamini-Hochberg procedure was used to control the FDR at 5%.Differences were considered statistically signif icant when the FDR corrected P value was<0.05.
Competing interests
The authors declare that they have no competing interests.
Authors’contributions
LZ conceived and designed the study,drafted the initial manuscript,and reviewed and revised the manuscript.ZG and GL conceptualized and designed the study,coordinated and supervised data collection,and critically commented on the important intellectual content of the manuscript.GPZ and YJ,critically reviewed and revised the manuscript.NL,DZ,YL,and LHZ,designed the clinical settings,went through ethic evaluation process,performed consents and questionnaire data sheets with patients,collected clinical samples and patients'information,and reviewed and revised the manuscript.JJY,YW,CL,JZ,BC,CZ,JW,and GYZ designed the data collection instruments,collected data,carried out the initial analyses,and reviewed and revised the manuscript.All authors approved the submission of the f inal manuscript and agreed to be responsible for all aspects of the work.
Acknowledgments
All phases of this study were supported by the National Natural Science Foundation of China(Grant No.81671362),the Medical and Health Science and Technology Development Projects of Shandong Province(Grant No.2015WSA01023),and Shandong Provincial Key Research and Development Program(Grant No.2018CXGC1219)to ZG;the National Natural Science Foundation of China(Grant No.31471202)and the Shandong Provincial Key Research and Development Program(Grant No.2016YYSP009),and the City of Weihai Technique Extension Project(Grant No.2016GNS023)to LZ.LZ is also supported by the Taishan Scholars Program of Shandong Province(Grant No.tshw20120206),China.
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.gpb.2019.01.002.
 Genomics,Proteomics & Bioinformatics2019年1期
Genomics,Proteomics & Bioinformatics2019年1期
- Genomics,Proteomics & Bioinformatics的其它文章
- Acknowledgments to Reviewers 2018
- GPA:A Microbial Genetic Polymorphisms Assignments Tool in Metagenomic Analysis by Bayesian Estimation
- How Microbes Shape Their Communities A Microbial Community Model Based on Functional Genes
- Agricultural Risk Factors Inf luence Microbial Ecology in Honghu Lake
- Inulin Can Alleviate Metabolism Disorders in ob/ob Miceby Partially Restoring Leptinrelated Pathways Mediated by Gut Microbiota
- Eあects of Proton Pump Inhibitors on the Gastrointestinal Microbiota in Gastroesophageal Ref lux Disease