
利用CRISPR/Cpf1系统构建人ABCG1和ABCG4基因敲除的293T细胞株
2019-05-14 11:22覃鸿妮谢钰珍蔡一林
西南农业学报 2019年4期
覃鸿妮,谢钰珍,马 傲,蔡一林
(1. 苏州工业园区服务外包职业学院,江苏 苏州 215123;2.金唯智生物科技有限公司,江苏 苏州 215123;3. 西南大学玉米研究所,重庆 400716)
【研究意义】三磷酸腺苷结合盒转运蛋白(ATP-binding cassette, ABC)超家族含有1个三磷酸腺苷(ATP)结合盒,可通过利用ATP水解过程释放的能量在溶质中跨膜转运各种生物分子[1]。ABC转运蛋白在脑组织中胆固醇的膜外转运和维持稳态中起着重要的调节作用。细胞内胆固醇主要通过SR-BI介导的双向转运模式,主动扩散以及由ABC转运蛋白介导的胆固醇单向跨膜转运机制排出胞外[2-3]。ABC转运蛋白根据其结构域和氨基酸同源性可分为ABCA~ABCG亚家族7个亚家族,人类基因组共包含48个具有转录活性的ABC转运蛋白转运子基因。ABCG1和ABCG4均为ABC 转运体超家族的成员。【前人研究进展】研究表明,ABCG1基因可广泛表达于各种人和巨噬细胞及小鼠组织中,能有效调节神经胆固醇的流动。Northern印迹分析显示人ABCG4 mRNA仅在大脑和眼睛的神经视网膜中高度表达,且ABCG4和ABCG1共有74 %的氨基酸序列同源性,所以ABCG4也可以促进胆固醇流向高密度脂蛋白(HDL)。目前关于ABCG1和ABCG4的功能研究还存在诸多问题,例如,对于ABCG1在动脉粥样硬化(AS)发病过程中的确切作用机制仍存在争议;ABC家族成员之间的相互作用关系仍有待进一步研究;动物模型的实验结果应用于人时存在一定的局限性;ABCG4转运子在脑细胞的特殊定位和功能分析,特别是ABCG4转运子在脑脂质动态平衡与神经系统疾病方面的功能还有待进一步阐明[4]。2012 年起 CRISPR/Cas9 被改造为基因编辑工具,可高效、便捷地完成基因定点突变、片段的敲除或敲入等[5-6]。【本研究切入点】CRISPR/Cpf1系统是在CRISPR/Cas9系统的研究基础上,发现的一种新型基因编辑技术。Cpf1相对于Cas9具有更高的特异性,比CRISPR/Cas9更适用于精确的基因组编辑,因此其在疾病模型建立、抗癌药物研制、干细胞治疗等生物和医学领域具有广泛的应用潜力。如何将CRISPR/Cpf1技术应用于疾病治疗,以及由此引发的安全性和伦理问题,也都需要更加深入的研究和规则的制定加以解决[7-9]。【拟解决的关键问题】本研究利用CRISPR/Cpf1基因组编辑技术,建立成熟的CRISPR/Cpf1基因敲除系统,为同类或其他基因的敲除提供借鉴;通过实现对细胞中ABCG1和ABCG4基因敲除模型的建立,为进一步研究细胞中胆固醇代谢的调节作用提供帮助,从而加快相关的医学临床应用研究的进展。
1 材料与方法
1.1 材料
人胚肾细胞株(293T)购于协和大学 (美国) 细胞库,Crispr/Cpf1质粒, Ascpf1和Lbcpf1载、Top10感受态细胞由金唯智生物科技有限公司构建;5×pfu buffer(通用)、10×pfu buffer(通用)、1×稀释 buffer、PBS、pfu DNA聚合酶、TaqDNA聚合酶、T4连接酶均由由苏州金唯智公司试剂组配制;BsmB1(ESP31)限制性内切酶购自thermo公司;T7E1限制性核酸内切酶购自北京唯尚立德公司;FBS(胎牛血清)购自BI(Biological Industrics)公司;DMEM高糖培养基购自HYClone公司;DNA凝胶回收试剂盒、质粒小量抽提试剂盒均购自AXYGEN公司;细胞基因组DNA抽提试剂盒购自TIANGEN公司。
1.2 方法
1.2.1 sgRNA引物的设计与合成 使用NCBI数据库下载ABCG1和ABCG4基因组完整序列,使用CRISPR在线设计工具(http://chopchop.cbu.uib.no/),分别在ABCG1和ABCG4蛋白质编码区(CDS区)前7个外显子设计sgRNA,根据网站设计的结果,选取2对得分较高的序列,在其两端加上酶切位点,在每条sgRNA序列的F链5’端添加AAAA,R链的5’端添加AGAT, 目的是与AsCpf1/LbCpf1质粒在BsmB1(ESP31)酶切后形成的黏性末端互补(表1)。引物序列由金唯智生物科技有限公司合成。
1.2.2 载体的构建及鉴定 AsCpf1/LbCpf1载体为含U6启动子(启动SgRNA)表达载体,其本身可以表达Cpf1酶,并具有卡那青霉素和嘌呤霉素筛选抗性。将ABCG1和ABCG4的2对sgRNA寡核苷酸单链分别退火合成双链,用T4连接酶和BsmB1(ESP31)限制性内切酶将退火后的引物通过边切边连的方法连接到AsCpf1/LbCpf1载体,构建AsCpf1-ABCG1-sgRNA1、LbCpf1-ABCG1-sgRNA2、AsCpf1-ABCG4-sgRNA1和LbCpf1-ABCG4-sgRNA2重组质粒;取连接产物10 μl转化至Top10 F’感受态细胞中,在Kan抗性的LB平板上筛选阳性克隆,抽提质粒并送进位置生物科技有限公司测序,使用LkO-5(GACTATCATATGCTTACCGT)通用引物测序。

表1 ABCG1和ABCG4的sgRNA寡核苷酸序列
注:黑色加粗字体表示在序列两端添加的碱基。
Note:The black bold font indicates the base added at both ends of the sequence.
1.2.3 细胞培养和细胞转染 提前将10 % FBS DMEM于水浴锅中37 ℃水浴10 min左右,293T细胞用1 mL培养基重悬,加入10 cm培养皿中,补10 mL DMEM培养基于5 % CO2,37 ℃恒温培养。细胞传两代后,进行计数,待细胞长到85 %以上进行铺板。细胞转染前24 h,进行计数按照一定比例铺6孔板。选择形态规则、融合度约在70 %~80 %的细胞,进行PEI质粒转染。将待转染的质粒(3000 ng)与PEI(2000 ng/mL)按3∶1的比例加入2 mL EP管,孵育5 min后,加入到NaCI和PEI的混合液中,混匀后静置20 min,转染293T细胞于5 % CO2,37 ℃恒温培养。重组质粒转染6 h后,用PBS或0.9 % NaCl清洗一次,培养基换成含10 % FBS培养细胞。转染48 h后,观察质粒转染细胞的转染效率,选取转染效率高的且形态规则的细胞进行加嘌呤霉素处理(终浓度20 μg/mL), 24~48 h后用0.9 % NaCl清洗换液,10 %FBS含双抗培养基培养细胞。
1.2.4 靶位点编辑效率验证 转染72 h后提取细胞基因组DNA,通过PCR扩增目的细胞基因组以得到目的DNA片段,回收PCR产物,采用T7E1核酸内切酶酶切回收后的AsCpf1-ABCG1-sgRNA1、LbCpf1-ABCG1-sgRNA2、AsCpf1-ABCG4-sgRNA1和LbCpf1-ABCG4-sgRNA2 DNA条带片段,使用3 %的琼脂糖凝胶检测DNA 片段,目的片段测序验证sgRNA编辑效率。
1.2.5 筛选稳定细胞株 转染96 h后,观察细胞生长状况并计数,采用有限稀释法按每孔5~6个细胞稀释到96孔板中,培养5~7 d后,在显微镜下观察96孔板中的细群落的个数,并做好记录。细胞生长到一定阶段会发生细胞堆积,根据细胞生长情况选取“单细胞”的96孔,可消化后缓慢吹打孔中的细胞,使细胞密度均匀,有利于加速细胞增殖的速度。5~7 d后,选取其中状态较好且已长满的96孔的单细胞系,经过消化后,取出一部分细胞液,剩余细胞液添加培养基继续培养。取出的细胞12 000 r/min 离心2 min,移去上清,PBS清洗1次,加入1×PCR稀释液裂解细胞2 U,蛋白酶K消化产物中蛋白,95 ℃ 5 min后,取5~10 μl作为巢式PCR扩增的模板。
1.2.6 单克隆细胞系验证 单克隆细胞基因组扩增产物切胶纯化后双向测序。对测序结果进行分析,选择正确有编辑的结果分别将其对应的PCR产物进行TA克隆,每个选取30个阳性克隆送测序。观察PCR产物TA克隆测序结果是否存在双克隆,如果存在需将双克隆菌液划线培养,挑斑并选取10个孔菌液送测,以进一步确定是否为单克隆细胞。根据测序结果,选取移码的突变细胞株转入6孔细胞培养皿进行扩大培养后抽提基因组,再次进行PCR扩增和产物纯化测序,确定为只含两种峰型的突变细胞株,将验证后的细胞转移到10 cm细胞皿扩大培养,待细胞长满后进行冻存。
2 结果与分析
2.1 重组质粒的测序
重组质粒AsCpf1-ABCG1-sgRNA1、LbCpf1-ABCG1-sgRNA2、AsCpf1-ABCG4-sgRNA1和LbCpf1-ABCG4-sgRNA2的测序结果显示,在靶位点插入的片段位置、方向及序列与预期结果一致,证明ABCG1和ABCG4的sgRNA插入到AsCpf1/LbCpf1载体中,重组质粒构建成功(图2~5)。
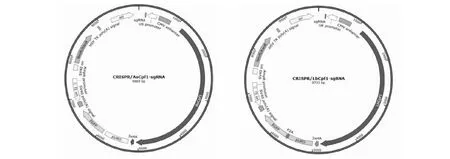
图1 CRISPR/Cpf1-sgRNA载体图谱Fig.1 CRISPR/Cpf1-sgRNA vector

图2 AsCpf1-ABCG1-sgRNA1靶位点测序结果Fig.2 Sequencing results of target sites of AsCpf1-ABCG1-sgRNA1

图3 LbCpf1-ABCG1-sgRNA2靶位点测序结果Fig.3 Sequencing results of LbCpf1-ABCG1-sgRNA2 target sites

图4 AsCpf1-ABCG4-sgRNA1靶位点测序结果Fig.4 Sequencing results of target sites of AsCpf1-ABCG4-sgRNA1

图5 LbCpf1-ABCG4-sgRNA2靶位点测序结果Fig.5 Sequencing results of LbCpf1-ABCG4-sgRNA2 target sites
2.2 细胞培养和细胞转染
将重组质粒转染至293T细胞,48 h后,荧光显微镜下观察4组293T细胞都有绿色荧光蛋白表达(图6),对比白光下的细胞对比明显较少。
2.3 sgRNA靶向敲除效率T7E1酶切验证
转染72 h后提取细胞基因组DNA,通过PCR扩增目的细胞基因组以得到目的DNA片段,AsCpf1-ABCG4-sgRNA 1细胞基因组没有扩增出来,其他3个靶位点均扩增出了目的条带,PCR产物经过T7E1酶切后,通过反显图可以看出产生明显的掉带,AsCpf1-ABCG1-sgRNA 1和LbCpf1-ABCG1-sgRNA 2掉带大小均为667 bp,AsCpf1-ABCG4-sgRNA 1掉落2个分别为220和447 bp的条带;LbCpf1-ABCG4-sgRNA 2掉带大小均为642 bp,LbCpf1-ABCG4-sgRNA 2掉落2个分别为156和486 bp的条带(图7),表明3个sgRNA靶向敲除效率均较高。将T7E1酶切验证有编辑效率的3个靶位点对应的PCR产物测序进一步验证,表明AsCpf1-ABCG1-sgRNA 1、LbCpf1-ABCG1-sgRNA 2和LbCpf1-ABCG4-sgRNA 2测序产物完全正确且均靶位点处有明显套峰,说明3个靶位点编辑成功。
2.4 稳定细胞株构建
将编辑后的细胞采用有限稀释法每孔4~5个分到96孔培养皿中,筛选并标记单克隆细胞(图8),对筛选结果进行统计分析,AsCpf1-ABCG1-sgRNA 1一共筛出10个单克隆细胞,LbCpf1-ABCG1-sgRNA 2一共筛出17个单克隆细胞,LbCpf1-ABCG4-sgRNA 2一共筛出8个单克隆细胞。
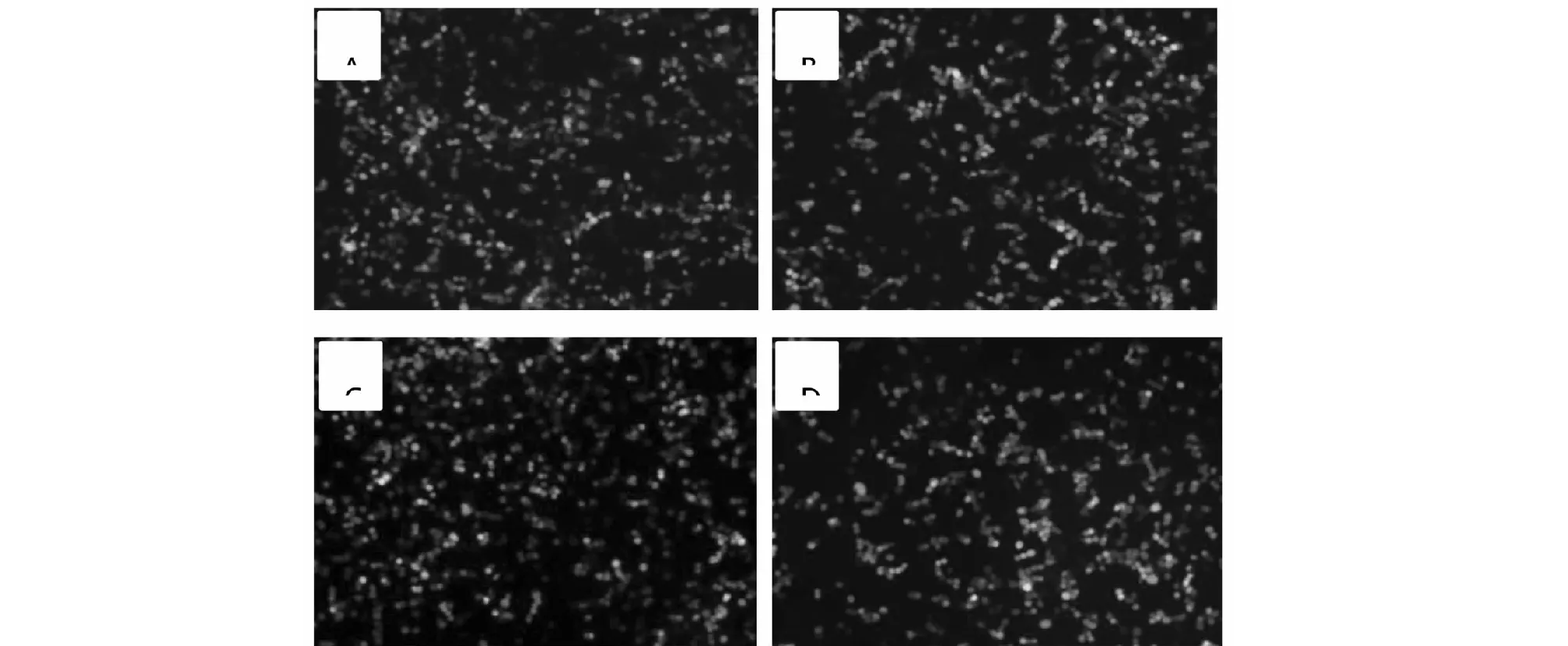
A:AsCpf1-ABCG1-sgRNA 1;B:LbCpf1-ABCG1-sgRNA 2;C:AsCpf1-ABCG4-sgRNA 1;D:AsCpf1-ABCG4-sgRNA 1图6 293T细胞荧光蛋白表达Fig.6 Fluorescent protein expression in 293T cells

Lane1:AsCpf1-1-g1产物+H2O;Lane2:AsCpf1-1-g1产物+T7E1酶;Lane3:LbCpf1-1-g2产物+H2O;Lane4:LbCpf1-1-g2产物+T7E1酶;Lane5:LbCpf1-4-g2产物+H2O;Lane6:LbCpf1-4-g2产物+7E1酶;LaneM:Marker DS5000图7 sgRNA靶向敲除效率Fig.7 Targeted knockout efficiency of sgRNA

A-单细胞在白光视野下(4×);B-单细胞在白光视野下(10×)A-single cell under white light (4×) and B-single cell under white light (10×)图8 单克隆细胞Fig.8 Monoclonal cells
2.5 单克隆细胞系编辑效率验证
单细胞系巢式PCR扩增送测,测序结果表明AsCpf1-ABCG1-sgRNA 1有编辑效率的有4个,LbCpf1-ABCG1-sgRNA 2有编辑效率的有3个,LbCpf1-ABCG4-sgRNA 2有编辑效率的有1个。将有编辑效率的细胞基因组PCR扩增产物进行TA克隆,挑取30个阳性克隆测序验证,其中5个单细胞系均在两套峰型以上,确定为双克隆细胞株,只有3个具有一套或两套峰型:
AsCpf1-ABCG1-sgRNA 1-1为单一的两套峰型,一套缺失7个碱基GACCTGC,一套缺失34个碱基ACATGGAGGCCACTGAGACGGACCTGCTGAATGG(图9),是纯合的单克隆细胞株;AsCpf1-ABCG1-sgRNA 1-2不是单一的两套峰型,一套缺失21个碱基GAGGCCACTGAGACGGACCTG,一套缺失25个碱基TGAGACCTGCTGAATGGACATCTGA(图10),有双克隆需进一步策序验证;LbCpf1-ABCG4-sgRNA 2-1仅为单一的一套峰型,缺失1个G碱基,突变7个碱基CGGCAGG(图11),是一个纯合的单克隆细胞株。
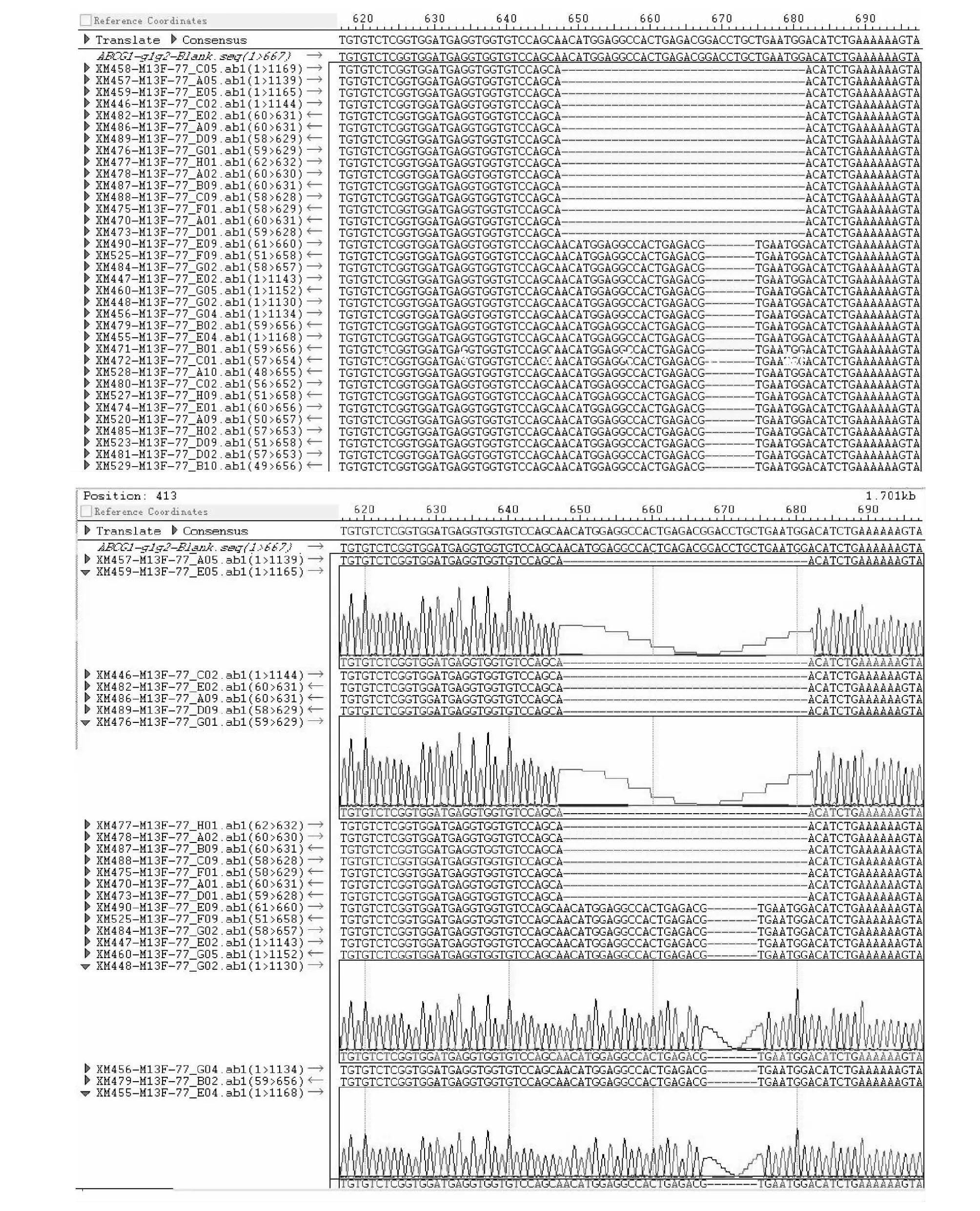
图9 AsCpf1-ABCG1-g1-1 TA克隆测序结果Fig.9 Cloning and sequencing results of AsCpf1-ABCG1-g1-1TA
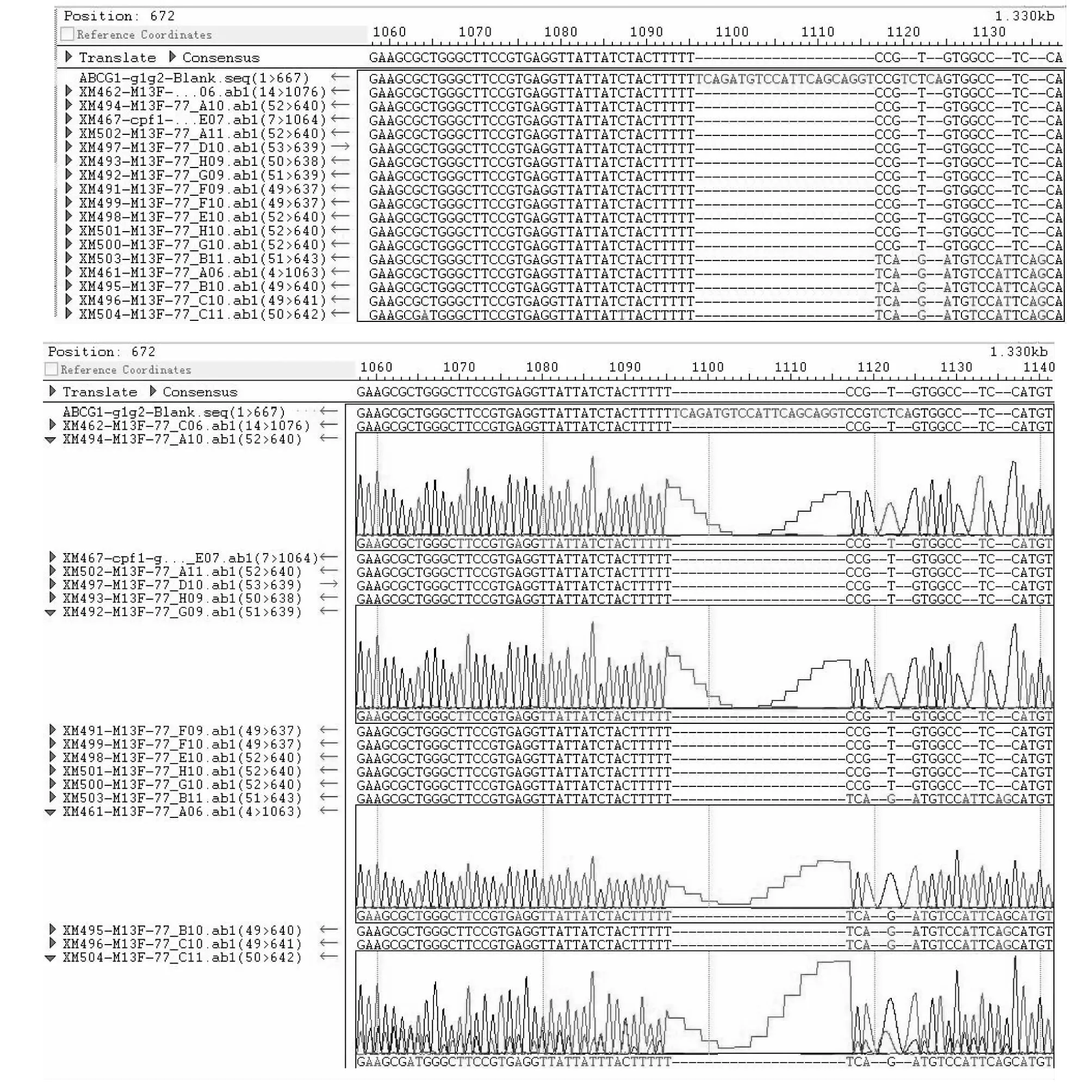
图10 AsCpf1-ABCG1-g1-2 TA克隆测序结果Fig.10 Cloning and sequencing results of AsCpf1-ABCG1-g1-2TA
2.6 双克隆细胞系编辑效率验证
将AsCpf1-ABCG1-sgRNA 1-2测序菌液XM504划线培养,选取10个孔菌液送测,分析其测序结果发现除了有两套目的峰型外,还有3个双克隆(图12)。通过对其峰型进行分析发现,双克隆的套峰是由GGCCACACGG和GGACATCTGA两种峰型杂合形成的,与两套目的峰型的结果一致,确定为双克隆细胞株。
2.7 单克隆细胞株测序作最终效率验证
根据TA克隆测序结果,选取移码的突变细胞株转入6孔细胞培养皿进行扩大培养后抽提基因组,再次进行PCR扩增和产物纯化并测序,通过分析比对靶位点附近的碱基序列及峰型,AsCpf1-ABCG1-sgRNA 1-1在靶位点处的套峰是由TGTCCAGCA和CGTGAATGG两种峰型杂合形成的,与TA克隆测序的结果一致,是一个纯合的单克隆细胞株(图13);AsCpf1-ABCG1-sgRNA 1-2在靶位点处的套峰是由GGCCACACGG和GGACATCTGA两种峰型杂合形成的,与TA克隆测序的结果一致,为只含两种突变类型的突变细胞株(图14),结合TA克隆和终验证测序结果发现靶位点后面有单个碱基的随机突变,不是稳定的单克隆细胞;LbCpf1-ABCG4-sgRNA 2-1靶位点处仅为1个碱基缺失,7个碱基突变(CGGCAGG)的单一峰型,是一个纯合的单克隆细胞株(图15)。

图11 AsCpf1-ABCG4-g2-1 TA克隆测序结果Fig.11 Cloning and sequencing results of AsCpf1-ABCG4-g2-1TA

图12 AsCpf1-ABCG1-g1-2 双克隆测序验证Fig.12 Verification of AsCpf1-ABCG1-g1-2 biclonal sequencing

图13 AsCpf1-ABCG1-g1-1终验证Fig.13 Verification of AsCpf1-ABCG1-g1-1

图14 AsCpf1-ABCG1-g1-2终验证Fig.14 Verification of AsCpf1-ABCG1-g1-2
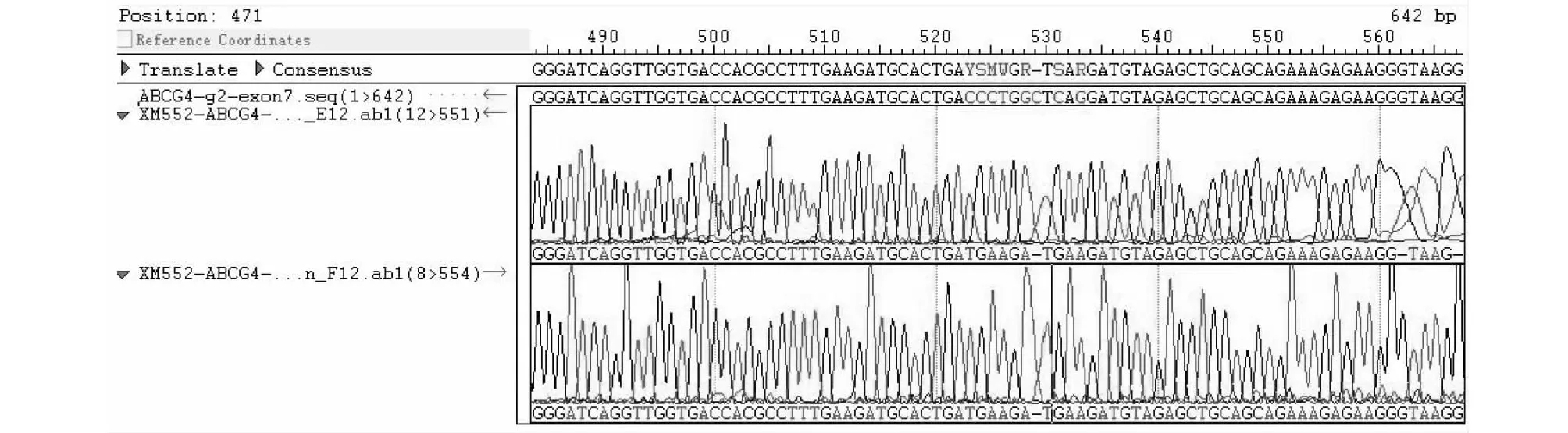
图15 AsCpf1-ABCG4-g2-1终验证Fig.15 Verification of AsCpf1-ABCG4-g2-1
3 讨 论
2012年,Jinek等证实了Cas9 可以在体外条件下与DNA 靶序列的特定位点结合并进行剪切,由此揭开了将CRISPR/Cas9 改造为基因编辑工具的序幕。近5 年来,CRISPR/Cas9技术为生物领域带来了巨大的冲击,2013 年被Science 杂志评为十大科学突破之一,2015 年更是荣登榜首。然而,随着研究的不断深入,CRISPR/Cas9暴露了它的缺陷和局限性,例如严重的脱靶效应。如何改造CRISPR/Cas9,降低其脱靶效应,提高基因编辑精确度,是目前科学家们关注和研究的重点[5]。
CRISPR/Cpf1 是继CRISPR/Cas9后,2015 年报道的一种新型CRISPR 效应蛋白,利用人工设计的sgRNA介导外源表达的Cpf1蛋白与基因组靶点特异性结合以实现对基因组DNA 的特异性切割,切割后的基因组DNA通过非同源末端连接(NHEJ)或同源重组(HDR)的方式进行修复,NHEJ是真核细胞中断裂DNA主要的修复方式,在修复的过程中会在断裂DNA链中随机的插入或缺失不定数目的碱基,通过基因移码突变来实现目的基因功能的缺失。CRISPR/Cpf1有利于克服CRISPR/Cas9 应用中的一些限制,作为新一代的CRISPR 基因组编辑工具,对CRISPR/Cpf1的研究才刚刚开始,有许多发展、改造和利用的空间[10]。
本研究一方面通过摸索条件,建立成熟的CRISPR/Cpf1基因敲除系统,为同类或其他基因的敲除提供借鉴。为了保证此次实验筛选的细胞为有编辑的单克隆细胞,在筛选的前期过程中,加入了嘌呤霉素大量杀死没有转染成功的细胞以提高细胞的编辑效率,在筛选后期过程中,对TA克隆测序中可能存在的双克隆细胞,进行双克隆菌液划线培养后进一步测序验证;另一方面,继CRISPR/Cpf1出现两年以来,各国研究组先后利用CRISPR/Cpf1 完成了对少数微生物、植物和动物基因组的编辑,目前还尚未有ABCG1/ABCG4基因采用CRISPR/Cpf1技术敲除的研究报道,ABCG1和ABCG4基因能够直接或间接的调控细胞中胆固醇代谢,深入研究其在胆固醇代谢中具体调节作用,将有助于了解一系列固醇类疾病的发病机制,并为此类疾病的防治提供新的方向与方法。本研究构建的目的细胞ABCG1/ABCG4基因敲除的细胞系,是深入研究ABCG1和ABCG4基因功能的前提基础。
4 结 论
本研究通过利用新型基因编辑技术CRISPR/Cpf1系统,利用人工设计的sgRNA介导外源表达的Cpf1蛋白与基因组靶点ABCG1和ABCG4特异性结合,通过基因移码突变来实现目的基因功能的缺失,永久性敲除了目的细胞靶基因,获得敲除ABCG1和ABCG4基因的稳定细胞株各1株(AsCpf1-ABCG1-sgRNA 1-1和LbCpf1-ABCG4-sgRNA 2-1)。稳定的基因敲除细胞株可直接用于后续研究,为进一步探索和证实ABCG1和ABCG4在细胞中胆固醇代谢和调节作用提供帮助,加快相关的医学临床应用研究的进展,同时建立了成熟的CRISPR/Cpf1基因敲除系统,可为同类或其他基因的敲除提供借鉴。
猜你喜欢
环球时报(2022-09-20)2022-09-20
昆明医科大学学报(2021年8期)2021-08-13
昆明医科大学学报(2021年3期)2021-07-22
昆明医科大学学报(2021年2期)2021-03-29
今日农业(2020年24期)2020-12-15
中西医结合肝病杂志(2020年2期)2020-10-27
医药前沿(2019年7期)2019-01-05
小资CHIC!ELEGANCE(2015年14期)2015-09-23
小资CHIC!ELEGANCE(2015年15期)2015-09-01
医学研究杂志(2015年9期)2015-07-01
