
CH3COO-与Ca2+的络合效应对方解石溶蚀的影响
2019-05-13 08:38:56李美蓉李晨初孙秀婷操应长
中国石油大学学报(自然科学版) 2019年2期
李美蓉, 安 波, 李晨初, 孙秀婷, 操应长
(1.中国石油大学(华东)理学院,山东青岛 266580; 2.中国石油大学(华东)化学工程学院,山东青岛 266580; 3.中国石油大学(华东)地球科学与技术学院,山东青岛 266580)
深埋藏条件下的碳酸盐岩溶蚀过程模拟[1-6]中,方解石的溶解过程非常关键,烃源岩裂解的关键组分是乙酸[7],研究乙酸根与钙离子的络合效应有助于解释乙酸对方解石的溶蚀规律及反应机制。Teng[8]观察方解石表面被乙酸侵蚀所形成的溶蚀特征后,提出“溶蚀窗”的概念;Fredd等[9]用方解石颗粒和乙酸水溶液在不同的地质条件开展实验,证明CH3COO-与Ca2+的络合作用有助于矿物的溶解;杨俊杰等[10]和崔振昂等[11]的实验结果表明,方解石的溶解过程受化学动力学控制,溶蚀量随温度的升高而减小;黄思静等[12]和佘敏等[13]认为,方解石的溶蚀过程受化学热力学控制,溶蚀量随温度的升高而增大。对于络合反应大都通过实施对照实验对其做定性描述[14-18],笔者从量子化学的角度对络合反应进行能量计算,将微观反应与宏观能量有机地联系起来,以乙酸溶蚀方解石过程中的固-液相分析为基础,在静态条件下探究反应温度对矿物溶蚀量的影响,利用Gaussian软件从量子化学的角度模拟计算络合反应的结合能,分析CH3COO-与Ca2+的络合效应对方解石溶蚀的影响。通过观测方解石解理面上次生孔—带—锥的演变过程,探讨方解石储层的形成机制。
1 实 验
1.1 实验材料和仪器
实验材料:方解石(中国石化胜利油田分析测试中心),分析纯乙酸(上海化学试剂有限公司),去离子水(实验室自制)。
仪器:电子天平(AL104,瑞士梅特勒科技有限公司),pH计(pHS-3C,上海雷磁有限公司),电热鼓风干燥箱(DHG-9023A,长春石油设备有限公司),水热反应釜(316L,山东恒化科技有限公司),原子吸收分光光度仪(HITACHIZ-500,日本日立科技有限公司),扫描电子显微镜(EM-30,韩国库赛姆科技有限公司),X射线光电子能谱(Agilent 5110,美国安捷伦技术有限公司),X射线晶粉衍射仪(X-Pert PRO,荷兰帕纳科技术有限公司)。
1.2 实验方法
将方解石在玛瑙研钵中粉碎,用筛网滤出粒径为0.250~0.297 mm的颗粒并将其浸泡在甲醇溶液中,利用超声波清洗至少3次,直到溶液完全干净,以确保颗粒表面吸附的极性物质和纳米级的粉末全部被清除。然后将矿物颗粒用去离子水反复冲洗3~5次,放入100 ℃的烘箱中干燥24 h。
静态溶蚀实验在400 cm3的水热反应釜中进行。准确称取3.0000 g 烘干的方解石粉末置于反应釜中,接着加入30 mmol/L的乙酸(pH=3.25)120.0 mL将其浸没。随后仔细密封容器并将其放入烘箱中加热至100 ℃,1 MPa。共设置9个实验,反应时间分别为0.5、1、2、3、4、5、7、9和11 h。每个实验设置3个平行,以保证每个样品都能按时足量取得。
实验结束后,待反应釜自然冷却至室温(时间至少6 h),用孔径为0.5 μm的滤纸过滤溶蚀液,将所得滤液汇集在干燥的烧杯中。先用注射器取样10 mL(用于原子吸收测试),再对剩余液体的pH值进行测定,经多次重复测量使pH值的偏差控制在±0.02以内。而滤出的粉末则与反应釜中残留的颗粒一起收集到新的烧杯中,直接放入100 ℃的烘箱中干燥24 h。
利用X射线光电子能谱(XPS)分析颗粒表面的元素含量,借助扫描电镜(SEM)观察固体表面的微观形貌,而样品中主要离子的浓度则用原子吸收分光光度仪进行测定。对于每组实验而言,测量值均要计算平均值和相对偏差,使结果的不确定性控制在±5%以内。
另外,将处理后的方解石粉末倒入研钵中持续研磨并不断过筛直至获得足量(质量约1.50 g)粒径小于40 μm的粉末。压片时,先将收集到的粉末用细筛过滤到显微镜载玻片中间的凹槽处,接着用小抹刀的刀口把粉末轻轻地压实,然后用载玻片的断口把多余或凸出的部分抹去,使得样品在窗口内分摊均匀,即得到一个可用于X射线衍射峰强度检测的较为平整的试片。
1.3 软件模拟
用Gaussian view 5.0 绘制出参与络合反应的各物质的结构,采用6-311+g(d,p) m062x基组,编写不同实验条件下的算法,然后运行Gaussian 09W对所设计的结构进行优化,从而得到该结构稳定存在时的基态能量。利用产物及反应物的基态能量之差(络合反应的结合能,其不同于反应的活化能,因为后者是用过渡态能量进行计算)来表征反应物之间络合能力的强弱,即ΔE结合能=E产物-∑E反应物。表1列出了不同温度下3种物质的基态能量。
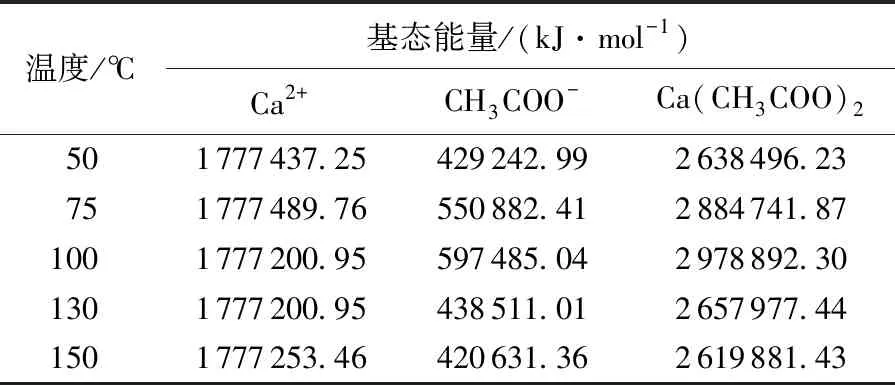
表1 不同温度下3种物质的基态能量Table 1 Ground state energy of three substances at different temperatures
2 结果分析
2.1 固 相
方解石表面的XRD图谱见图1。由图1可知,当扫描范围在26°~32°时,方解石的响应最为强烈,然而并未发现任何其他矿物明显的衍射峰,这说明反应后方解石表面没有次生矿物形成或者其生成量太少以至于仪器难以检测。随着反应的持续进行,方解石的衍射峰强度逐渐减弱,说明其与乙酸发生作用,以新的形式被转移到溶液中,使得其固相含量不断减少。反应3 h后,方解石的峰强度变化显著,说明乙酸对方解石的侵蚀作用较强,方解石的溶蚀量增多。反应5 h后,尽管峰强度继续减弱,但幅度变小,说明之前溶蚀反应的优势被削弱,方解石的溶蚀量减少,预示着溶蚀过程即将结束。
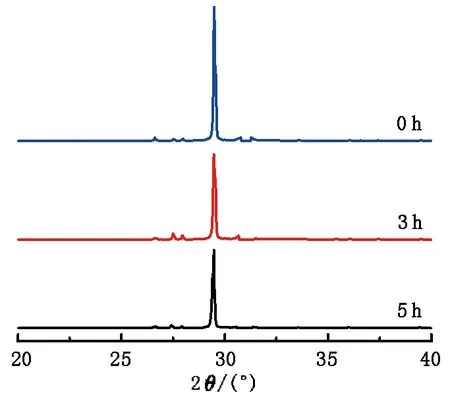
图1 方解石表面的XRD图谱Fig.1 XRD pattern of calcite surface
方解石表面的元素质量分数见图2。可以看出,反应前方解石表面钙元素的含量较多,而镁、铁元素的含量较少。随着反应时间的延长,钙元素的含量逐渐降低,其中前一阶段的减幅为18.47%,较后一阶段高8.86%,这说明前3 h内乙酸对方解石的溶蚀较为剧烈,固相中钙元素被大量转移,故推测此阶段为矿物溶蚀进程中的关键期。离子在溶液中的自由迁移,H+在矿物表面的置换作用以及有机酸阴离子与金属阳离子的络合效应,均在此最为明显。然而镁、铁元素的含量虽存在不同程度的减少,但其对矿物溶蚀量的贡献太小,可以忽略不计。
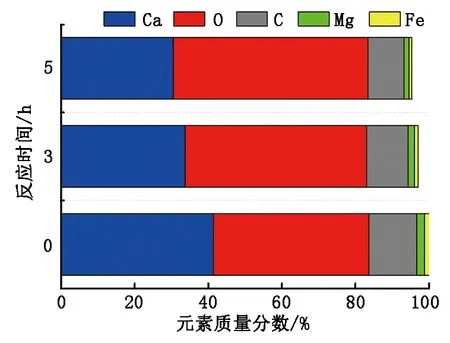
图2 方解石表面的元素含量Fig.2 Element content of calcite surface
2.2 液 相
对溶液中3种金属离子的质量摩尔浓度进行了测定,结果表明,Mg2+、Fe2+的质量摩尔浓度与Ca2+的不在同一个数量级,甚至低于仪器的最低检测限,以至于其质量摩尔浓度无法测定,Ca2+质量摩尔浓度和溶液pH值随时间的变化曲线见图3。其中k1、k2和k3分别为不同阶段的平均反应速率,mmol·kg-1·h-1(以Ca2+质量摩尔浓度计算)。

图3 Ca2+质量摩尔浓度以及溶液pH值随时间的变化Fig.3 Variation of Ca2+concentration and pH value of solution with time
由图3可知,方解石的溶蚀过程大致分为3个阶段:① 上升阶段,矿物表面的Ca2+与乙酸电离出的H+发生置换反应,被释放到溶液中,与此同时,溶液中的CH3COO-与Ca2+形成乙酸钙络合物,促进了矿物的溶解。Putnis等[19]提出溶液中存在少量的Mg2+能够起到催化作用,推动溶解反应的进行,而这其实是一种同离子效应。方解石溶解的过程中会放出少量CO2,而寿建峰等[20]和张少敏等[21]的实验证明体系中存在CO2能够增大方解石的溶解度,加剧溶液的侵蚀作用。反应3 h后,溶液中Ca2+的质量摩尔浓度已高达13.52 mmol/kg,继续反应2 h,其质量摩尔浓度增大了1.72 mmol/kg,说明方解石的溶解在前3 h内已近乎完全,从而进一步证明了固相分析中的猜测是合理的;② 过渡阶段,随着乙酸解离能力的逐渐减弱,溶液pH值慢慢增大,能够被置换出的Ca2+的数量也不断减少,矿物溶蚀进程受阻,Ca2+质量摩尔浓度的变化趋于平缓,平均反应速率仅为上一阶段的1/5,从而成为了反应由盛变衰的转折点;③稳定阶段,溶液呈近中性,H+几乎消耗殆尽,矿物的溶解过程难以持续下去,故Ca2+的溶出质量摩尔浓度基本保持不变。
测定50、75、130和150 ℃(压力均为1 MPa)下溶液中Ca2+质量摩尔浓度随反应时间的变化(表2)以及方解石溶蚀量随温度的变化(图4)。结果表明,在任一温度下,Ca2+质量摩尔浓度在初始3 h内急剧增大,历经2 h的缓慢增长之后,基本保持恒定直至反应结束,这与100 ℃下Ca2+质量摩尔浓度随时间的变化趋势高度吻合。

表2 不同温度下溶液中Ca2+质量摩尔浓度随时间的变化
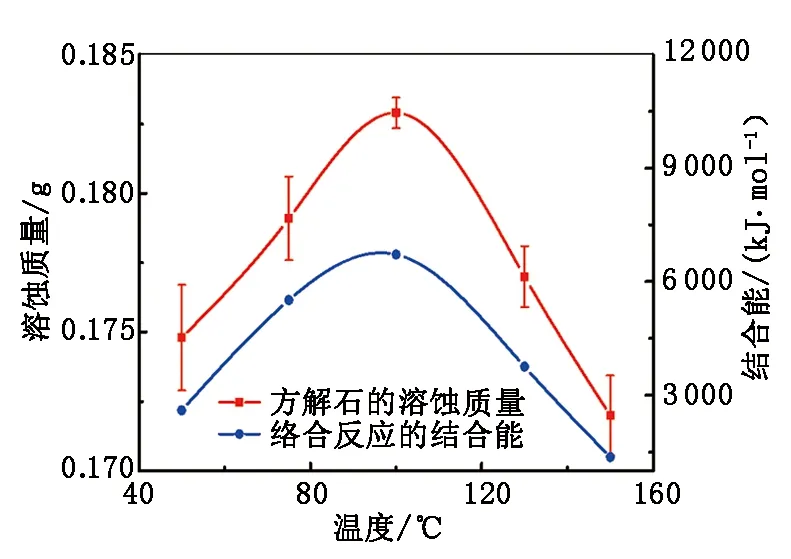
图4 不同温度下方解石的溶蚀质量以及络合 反应的结合能Fig.4 Dissolution of calcite and binding energy of complexing reaction at different temperatures
温度升高有助于提高反应速率以及加快溶液中离子的扩散,显然,溶液中Ca2+的最大溶出质量摩尔浓度应该随着温度的升高而增大,矿物的溶蚀量也应呈现相同的变化趋势。但通过分析表2中的数据可知,随着温度的升高,溶液中Ca2+的最终溶出质量摩尔浓度呈现先增加后减少的趋势,在100 ℃时达到最大值15.75 mmol/kg;对比不同温度下反应前后方解石的质量变化(图4)可知,溶蚀质量同样随着温度的上升呈现先增大后减小的变化,在100 ℃时达到了最大值0.182 9 g。该实验结果与理论分析并不一致,很可能是由于忽略了CH3COO-与Ca2+之间的相互作用所引起的,尽管众多研究学者普遍认为二者之间的络合效应所产生的影响远不及H+在矿物表面的置换作用,但若从量子化学的角度分析此问题,应用Gaussian分子模拟软件计算络合反应的结合能(衡量CH3COO-与Ca2+络合作用的强弱),却能够给出一种新的解释,即H+的交换作用、溶液中离子的自由扩散以及CH3COO-与Ca2+之间的络合效应共同承担了溶蚀矿物的任务,在100 ℃时,三者的综合作用得到了最大的发挥,使得乙酸对方解石的溶蚀最为剧烈。低于100 ℃时,络合反应的结合能随着温度的上升而增大,络合效应在整个溶蚀过程中的地位不断提升,更多被交换出的Ca2+以络合物的形式转移到溶液中,加快了H+与矿物表面阳离子置换的进程,从而进一步推动溶蚀反应的进行。高于100 ℃时,温度升高,络合反应的结合能减小,络合效应被削弱,侵蚀矿物的任务主要由前两者来继续完成。但随着溶液pH值的逐步增大,能用来交换的H+的数目愈来愈少,单凭溶液中离子的扩散作用已无法继续维持溶蚀反应的进行,故溶蚀过程被迫停止。在复杂的储层环境中,随着埋藏深度的增加,地层温度上升且乙酸浓度降低,使得方解石的溶蚀过程较为平和,所以深埋藏条件下有利于方解石储层的形成与发育。相较之下,浅埋藏条件下的地层温度较低且乙酸浓度较大,方解石的溶蚀过程变得愈发剧烈,扩大了储层内部的间隙,有利于油气的存储与转移。值得注意的是,杨俊杰等[10,12]是在动态条件下开展的模拟实验,引入了更多的不确定因素,如离子迁移速率,反应热损耗等,从而得到了不同的结果,结合本实验的分析,笔者认为后者的结论较为可靠。
2.3 方解石表面的微观形貌
反应后方解石表面的微观形貌见图5。经扫描电镜局部放大后,可以清晰地观察到方解石表面出现了多种明显的溶蚀现象,这是矿物发生选择性溶蚀的结果。

图5 反应后方解石表面的微观形貌Fig.5 Microscopic morphology of calcite surface after reaction
选择性溶蚀的实质即晶体表面的某些离子在极性分子电荷的影响下向溶液中逃逸,导致晶格原有的形态被破坏。首先解理面上会形成大量不规则的溶孔,随后其面积不断扩大且彼此间相互连接从而演变成条纹状的溶蚀带,将更多的表面暴露于溶蚀液中。然而CO32-与Ca2+在不同方向上的排列方式存在差异,造成解理面上的活性位点沿各个方向的溶解速率不同(但存在一定的比例关系且CO32-解离的速率较快),最终产生了溶蚀晶锥。不过它只是方解石解离面溶蚀过程中的一种阶段性产物,随着反应的进一步深入,晶锥的规模会逐渐变小甚至消失直至裸露出下方新的表面(图6,据孟繁奇等[17],有修改)。
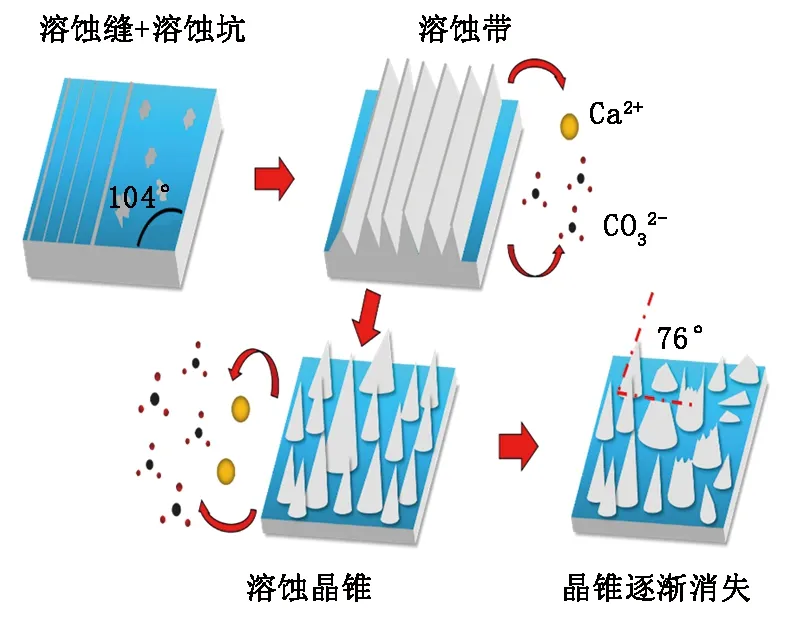
图6 方解石的溶蚀过程Fig.6 Dissolution process of calcite
3 结 论
(1)随着温度的升高,溶液中Ca2+的最终溶出质量摩尔浓度呈现先增加后减少的趋势,在100 ℃时达到了最大值;而方解石的溶蚀质量同样随着温度的上升呈现先增大后减小的变化,在100 ℃时达到了最大值。
(2)100 ℃时,CH3COO-与Ca2+之间的络合效应最强,与溶液中离子的自由扩散以及H+的交换作用相结合,使得乙酸对方解石的溶蚀最为剧烈。低于100 ℃时,络合反应的结合能随着温度的上升而增大,更多被交换出的Ca2+以络合物的形式转移到溶液中,进一步促进溶蚀反应的进行。高于100 ℃时,温度升高,络合反应的结合能减小,侵蚀矿物的任务主要由氢离子的置换作用和溶液中离子的自由扩散共同承担。然而随着溶液pH值的逐步增大,能用来交换的H+的数目不断减少,单凭溶液中离子的扩散作用已无法继续维持溶蚀反应的进行,因此溶蚀过程被迫停止。
(3)晶体表面上的CO32-与Ca2+在不同方向上的排列方式存在差异,造成解理面上的活性位点沿各个方向的溶解速率不同,最终导致溶蚀晶锥的形成。不过它只是方解石解理面溶蚀过程中的一种阶段性产物,随着反应的持续进行,晶锥的规模会逐渐变小甚至消失直至下方新的表面露出。
猜你喜欢
硅酸盐通报(2022年8期)2022-09-08 04:25:42
中学生数理化·高一版(2022年4期)2022-05-09 15:36:00
矿产综合利用(2021年3期)2021-07-14 03:46:54
河北画报(2020年10期)2020-11-26 07:20:56
矿产综合利用(2020年1期)2020-07-24 08:50:46
英美文学研究论丛(2018年2期)2018-08-27 01:56:44
浙江大学学报(工学版)(2016年9期)2016-06-05 09:20:57
当代化工研究(2016年5期)2016-03-20 16:21:29
中国非金属矿工业导刊(2014年4期)2014-02-28 09:20:51
江西理工大学学报(2013年1期)2013-03-20 14:57:07
