
铁电层厚度对(BiCoO3)n/(La2/3Sr1/3MnO3)1超晶格磁电性质的影响
2019-04-28 08:53郭根材明帮铭
原子与分子物理学报 2019年5期
张 威, 张 铭, 郭根材, 明帮铭
(北京工业大学材料科学与工程学院,北京100124)
1 引 言
多铁材料同时具有两种或两种以上的铁性有序,如铁磁性、铁电性和铁弹性. 在多铁材料中,铁磁性与铁电性之间相互耦合,通过电场可以改变磁化方向,反之,也可以通过磁场改变电极化方向[1, 2]. 这在多态存储器件和电场调控的自旋电子器件等方面具有潜在应用[3-5]. 近些年来,相比于单相多铁材料,作为异质结或超晶格结构的复合多铁材料具有更大的铁电极化值和更高的铁磁居里温度,因而受到人们的广泛关注[6-8]. 与此同时复合多铁材料还具有丰富的物理特性,如自旋运输特性、磁电耦合效应等[9, 10].
钙钛矿锰酸盐La2/3Sr1/3MnO3(LSMO)因其具有半金属性和极高的铁磁居里温度Tc约为370 K,在自旋电子学领域是极有前景的候选者之一[11]. 与LSMO构建异质结构通常表现出优异的物理性质,例如2010年Garcia等人在NdGaO3衬底上构建了Fe/BaTiO3/La0.67Sr0.33MnO3多铁异质结,实现了BaTiO3的铁电极化对异质结隧穿磁致电阻效应的调控[12]; Eerenstein等人发现了在LSMO/BaTiO3异质结中由界面应变引起的磁电耦合效应[13]. 利用铁磁材料与具有反铁磁性的多铁材料耦合时的交换偏置作用也可以实现磁电控制. Yu等人使用X射线磁性圆二色性法验证了LSMO/BiFeO3(BFO)界面处存在显著的交换偏置作用[14]. Wu等人证实了在LSMO/BFO异质结中电场能够对交换偏置进行调控[15]. Jilili等人发现LSMO/BFO超晶格仍然保持半金属特性[16]. BiCoO3与PbTiO3有相同的构型,具有比BiFeO3更大的理论铁电极化值170μC/cm2. 此外,已有实验通过中子衍射明确证实BCO具有低于奈尔温度Tn=470 K的长程C型反铁磁序[17, 18]. 在目前已知的多铁材料中, BiCoO3(BCO)被认为是最有希望的多铁材料候选者之一. 因此,同时具有极大铁电极化值和C型反铁磁序的BCO与LSMO构成超晶格很可能表现出新颖的磁电性质. 此外,铁电薄膜厚度对异质界面的物理性质起着很重要的作用[19-21]. 但是,当前对于多铁材料与LSMO所构成异质界面中多铁薄膜厚度的研究仍然较少. 在这项工作中,我们构建了BCO和LSMO组成的超晶格,并使用第一性原理计算理论研究了BCO侧厚度对(BCO)n/(LSMO)1超晶格((BCO)n/(LSMO)1表示BCO侧为n个单胞层)的电子结构、极性位移、磁性等的影响.
2 计算方法和模型
本文采用投影缀加平面波赝势法[22]和广义梯度近似(GGA)来执行密度泛函理论计算[23],该计算过程是在第一性原理计算软件包VASP[24]上完成的. 基于密度泛函理论的第一性原理计算方法已经成为现代材料计算和设计的重要基础,并已得到广泛的应用[25-27]. 考虑到本文中所研究的体系包含3d过渡族金属原子,其3d轨道电子具有强的库仑排斥作用,我们采取了加U计算的方法来进行处理. 根据对之前关于BCO和LSMO体材料的理论研究的参考,在本次计算过程中分别将对应Mn的U值设置为2 eV,对应Co的U值设置为5 eV[28, 29]. 在整个计算过程中,平面波基组的截断能量选用500eV. 以Gamma点为中心的7×7×7和7×7×1的k点网格分别应用于体材料和超晶格的计算. 自洽的能量收敛标准设定为10-5eV,结构优化的收敛标准设定为每个原子所受到的Hellmann-Feynman力小于0.01 eV/Å.
具有钙钛矿结构的四方相BCO的空间对称群为P4mm,其晶格常数为a=3.751 Å,c/a=1.283[18, 28]. 与此同时,它的磁性表现为C型反铁磁,即Co3+离子在xy平面呈反铁磁序排列且在z轴方向呈铁磁序排列. LSMO也是具有四方相的钙钛矿结构,其晶格常数a=3.880 Å,c/a=3.000[30]. 它的磁性表现为铁磁,即Mn3+离子在x轴、y轴、z轴三个方向均呈铁磁序排列. 在搭建超晶格模型时,我们选择将面内晶格常数设置为√2a×√2a,这样做的目的是为了能够在超晶格中呈现出BCO的C型反铁磁序. 如图1所示,一个LSMO的单胞是由LaO、SrO、MnAO2、MnBO2四层有序堆垛而成的,其中位于LaO层与SrO层之间的是MnA原子,位于两个LaO层之间的是MnB原子. 经计算我们发现BCO(001)与LSMO(001)的面内晶格常数相差在3.3%左右,具有良好的晶格匹配度(<5%). 在本文中我们选取LSMO侧为一个单胞(u.c.)层,BCO侧分别为1~4个单胞(u.c.)层,从而研究BCO层厚度对(BCO)n/(LSMO)1超晶格的电子结构和磁电性质的影响.
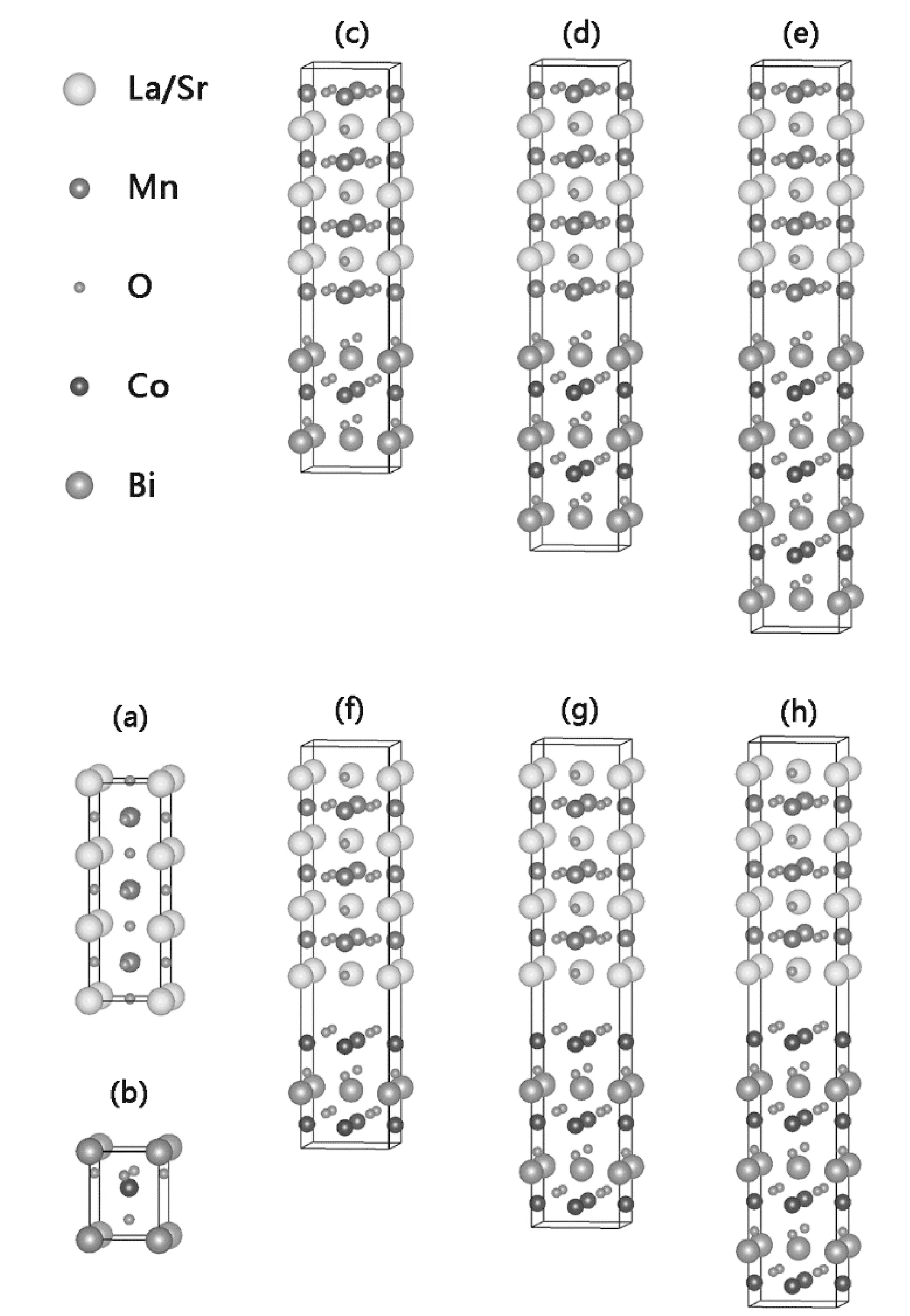
图1 (a)LSMO体材料的结构示意图;(b)BCO体材料的结构示意图;(c)-(e)BiO/MnBO 2超晶格的结构示意图,BCO侧分别为1-3个单胞层;(f)-(h)CoO2/LaO超晶格的结构示意图,BCO侧分别为1-3个单胞层. Figure 1 (a) Side view of schematic structures of unit cells for bulk LMSO, (b) bulk BCO; (c)-(e) BiO/MnBO2 superlattice, BCO side is 1-3 unit cells, respectively; (f)-(h) CoO2/LaO superlattice, BCO side is 1-3 unit cells, respectively.
3 结果与讨论
BCO与LSMO均为ABO3构型的钙钛矿结构,在(001)面内共有八种形式的截断面,分别为BCO的BiO、CoO2和LSMO的LaO、SrO、MnAO2、MnBO2. 一般情况下,钙钛矿结构通常是由AO、BO在[001]方向上以周期循环的方式堆垛而成. 因此以AO/BO方式堆垛的界面较AO/AO或BO/BO方式而言通常能量会相对较小,从之前关于钙钛矿的界面研究中同样也可以得出这样的规律[31, 32]. 于是在本文中我们只考虑了界面处为AO/BO的堆垛方式,分别搭建了以BiO和MnAO2、BiO和MnBO2、CoO2和LaO、CoO2和SrO为界面构型的超晶格,并对它们进行了研究. 文中我们通过计算BCO与LSMO之间的形成能来判断所搭建的超晶格在能量上的相对稳定性,Wsep=(ELSMO+EBCO-ELSMO/BCO)/2,其中EBCO/LSMO是异质结的总能量,EBCO和ELSMO分别代表BCO或LSMO具有相同结构的超晶格的能量.
图2分别展示了我们通过计算得到的结构完全弛豫之后BCO和LSMO体材料的态密度. 从计算结果来看, BCO的整体带隙为1.53 eV,与之前的理论计算结构保持一致,同时相比于实验得出的结果1.70 eV相对较小[18, 28]. 这是由于众所周知的DFT计算过程中交换关联泛函的导数不连续导致的[33]. 其中在xy面内相邻两个Co原子的自旋是反平行的,计算得到的Co原子磁矩为± 3.019μB. 这一结果也与之前报道过的理论研究保持一致,同时也和实验值3.24μB很好地吻合[34]. 从图2中可以出, LSMO呈现出半金属特性. 同时,通过计算得到的MnA、MnB的磁矩分别为3.43μB和3.52μB,很接近实验值[35]. 以上结果充分地证实了本文中所使用计算参数的可靠性.
在研究工作的前期阶段,我们首先对LSMO与BCO间的形成能进行了计算. 这里我们考虑了四种不同界面构型的超晶格. 经完全的结构弛豫后,形成能能量最低的是以CoO2和LaO为界面构型的超晶格,其次是界面构型为BiO/MnBO2的超晶格. 因此本文着重对这两个界面构型的超晶格中BCO侧厚度对其电子结构、极性位移等的影响进行了研究.
为了方便起见,在这项工作中我们分别使用BiO/MnBO2、BiO/MnAO2、CoO2/LaO、CoO2/SrO来表示这四种超晶格. 此外,将BCO铁电极化指向的界面用“+”表示,将BCO铁电极化背向的界面用“-”表示. 作为异质结的超晶格,它的电子结构很大程度上取决于其中界面处的耦合作用. 而其界面处的耦合作用很大程度上又取决于由结构弛豫引起的局部结构畸变,即界面处附近原子的位移. 因此我们在讨论时会更关注界面处的原子. 作为多铁材料, BCO本身具有很大的铁电极化值. 对于BCO区域, CoO2层、BiO层的相对位移在很大程度上反应了其铁电极化的大小. 从表1中,我们可以看出随BCO厚度的变化,超晶格中界面处的结构弛豫是明显不同的. 在BiO/MnBO2+、BiO/MnBO2-、CoO2/LaO+界面处,随BCO厚度的增加,BCO侧极性位移dz也不断变大. BiO/MnBO2+和BiO/MnBO2-界面处的极性位移dz在BCO增至两层时达到临界值0.965 Å、0.372 Å. CoO2/LaO+界面处的极性位移在BCO增至三层时达到临界值0.602 Å. 随着铁电层的厚度增加,其铁电极化值也会随之增大,达到临界厚度便不再变化. 而CoO2/LaO-界面处的极性位移在BCO增至三层时有一定的减小,由0.626 Å减小至0.547 Å. 关于超晶格的电子结构,我们计算并绘制了各超晶格的能带图,如图2所示. 来自BCO、LSMO部分的贡献,图中分别用蓝色和红色进行了标记. 在BiO/MnBO2超晶格中,BCO侧厚度为一个单胞层时,超晶格总的电子结构表现为半金属特性,如图2(a). 其中,上自旋电子在费米面处有填充;下自旋电子在费米面处没有占据且下自旋电子的带隙为1.08 eV. 随BCO厚度增加,达到两个单胞层以后,超晶格的电子结构由半金属性转变为金属性. 并且(BCO)2/(LSMO)1和(BCO)3/(LSMO)1超晶格的电子结构很类似. 这样的变化趋势与BiO/MnBO2超晶格界面处的极性位移相吻合. 值得指出的是,在BiO/MnBO2超晶格中,其电子结构尤其是自旋电子特性是可以通过铁电层厚度来进行调节的. 而在以CoO2、LaO作为界面的CoO2/LaO超晶格中,无论BCO厚度如何变化,超晶格总的电子结构均表现为金属特性. 作为半金属材料的LSMO与作为绝缘体的BCO在形成超晶格后表现为金属性. 这些现象充分地说明了超晶格的物理性质很大程度上取决于两个不同材料之间的界面耦合作用[36]. 最后我们计算了超晶格中界面处Co原子的自旋极化率(这里定义P= (N↑(EF) -N↓(EF))/(N↑(EF) +N↓(EF)),其中N↑(EF)和N↓(EF)分别代表态密度图中费米能级处上自旋和下自旋的值). 各界面处的自旋极化率的大小不尽相同,如表2. 在BiO/MnBO2+、BiO/MnBO2-和CoO2/LaO+界面处Co原子的自旋极化率大小变化趋势基本一致,它们的自旋极化率的大小均随BCO厚度增加不断减小,分别由100%到0%、由100%到-23.44%和-81.82%到-2.96%. 在CoO2/LaO-界面处,Co原子的自旋极化率在BCO厚度为1至4个单胞层时,分别为12.09%、0%、-97.18%、85.06%. 从界面处自旋极化率的角度看,超晶格中大部分界面同样表现出受BCO厚度变化的影响趋势. 而在CoO2/LaO-界面处极性位移、自旋极化率等没有随BCO厚度表现出规律性变化,这一现象仍值得进一步的研究.
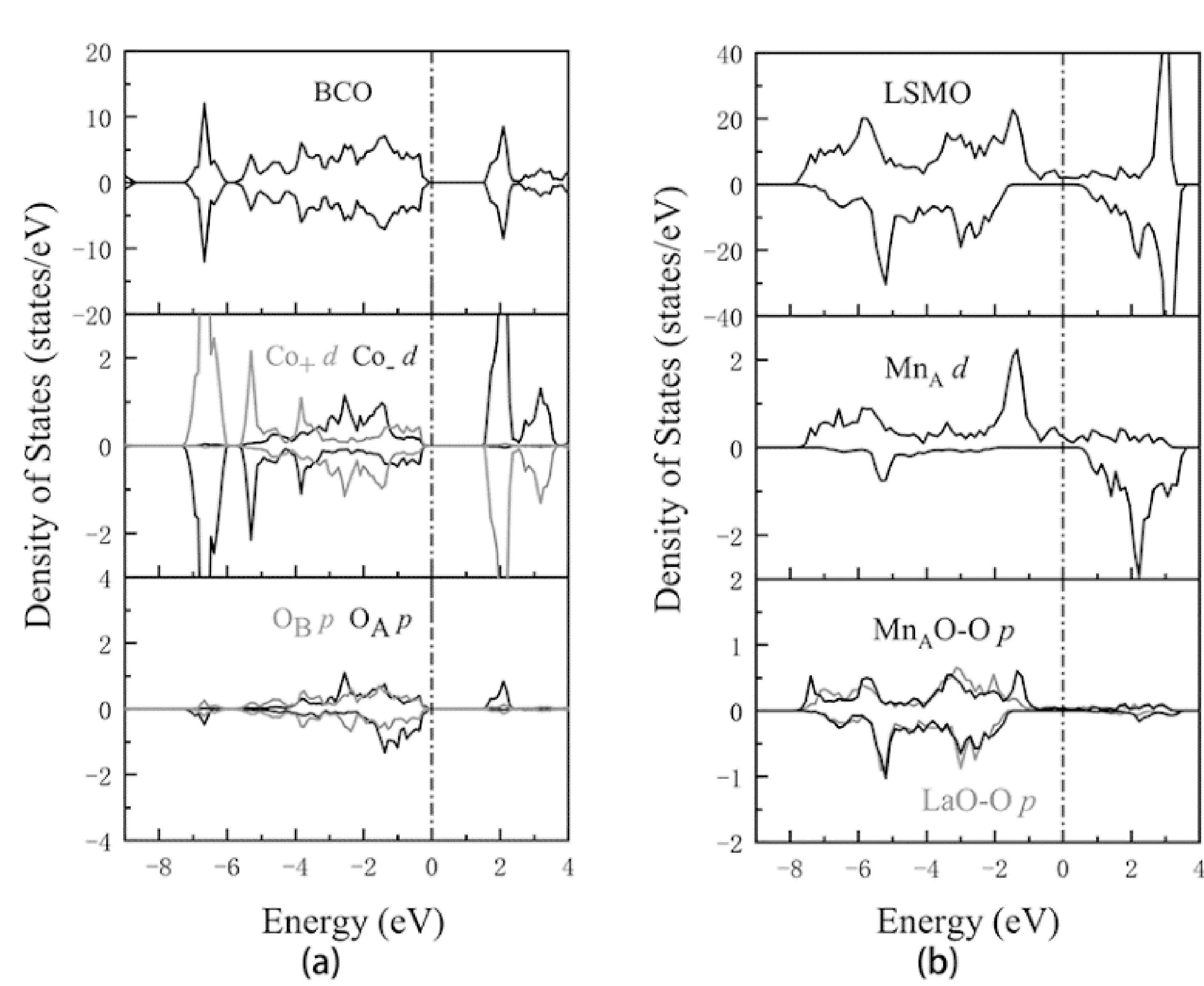
图2 (a)-(c)BCO体材料的总态密度和局部态密度;(d)-(f)LSMO体材料的总态密度和局部态密度. Co+和Co-代表具有不同反铁磁取向的Co原子. 费米能级设为零点,并以点划线标出. Figure 2 (a)-(c) Total and partial DOS (PDOS) for bulk BCO; (e)-(f) Total and partial DOS for bulk LSMO. Co+ and Co- represent the antiferromagnetic aligned Co ions. The Fermi energy is set to zero and depicted by a vertical dot line.
表1 BiO/MnBO2、CoO2/LaO超晶格中界面处的极性位移dz随BCO侧厚度的变化. (单位:Å)
Table 1 The polarity displacementdzat the interface of the BiO/MnBO2, CoO2/LaO superlattice, BCO side is 1-4 unit cells (u.c.), respectively. (Unit: Å)
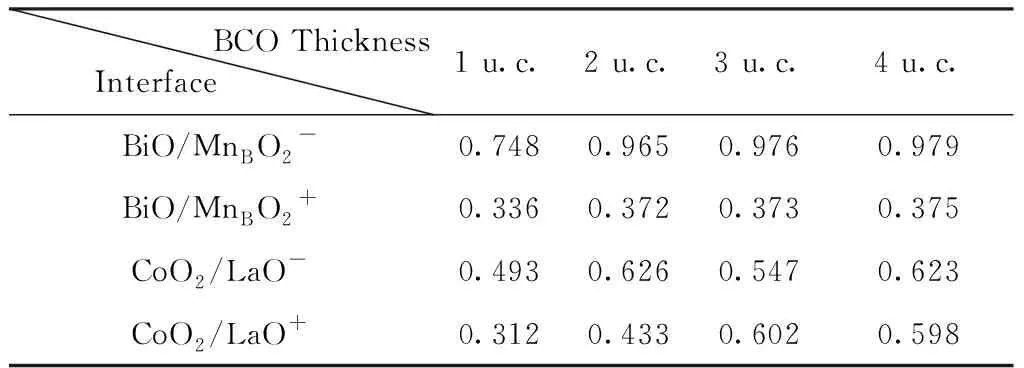
BCO ThicknessInterface 1 u.c.2 u.c.3 u.c.4 u.c.BiO/MnBO2-0.7480.9650.9760.979BiO/MnBO2+0.3360.3720.3730.375CoO2/LaO-0.4930.6260.5470.623CoO2/LaO+0.3120.4330.6020.598
表2 BiO/MnBO2、CoO2/LaO超晶格中界面处的Co原子自旋极化率P随BCO侧厚度的变化. (单位:%)
Table 2 The Co atom’s spin polarizationPat the interface of the BiO/MnBO2, CoO2/LaO superlattice, BCO side is 1-4 unit cells, respectively. (Unit: %)

BCO ThicknessInterface 1 u.c.2 u.c.3 u.c.4 u.c.BiO/MnBO2-1009.0900BiO/MnBO2+100-7.98-4.62-23.44CoO2/LaO-12.090-97.1885.06CoO2/LaO+-81.82-28.53-2.94-2.96
4 结 论
本文通过基于密度泛函理论的第一性原理计算方法对结构较稳定的(BCO)n/(LSMO)1超晶格的原子结构、电子结构和界面处自旋极化率以及BCO侧薄膜厚度对它们的影响进行了分析研究. 我们发现在BiO/MnBO2中,当BCO侧为一个单胞层时,超晶格的能带结构表现为半金属性. 其余超晶格的能带结构均表现为金属性. 另外,随BCO的厚度增加,界面处的极性位移逐渐减小至临界值,界面处Co原子自旋极化率逐渐趋近于0%. 这些研究结果表明,铁电层厚度是影响多铁界面磁电性质的重要因素.
猜你喜欢
数学物理学报(2022年5期)2022-10-09
当代作家(2021年11期)2021-12-17
北方论丛(2021年2期)2021-05-22
科学(2020年4期)2020-11-26
科学(2020年4期)2020-01-11
数学物理学报(2019年5期)2019-11-29
黑龙江电力(2017年1期)2017-05-17
学生天地·小学低年级版(2016年9期)2016-05-14
云南师范大学学报(自然科学版)(2015年5期)2015-12-26
中央民族大学学报(自然科学版)(2015年2期)2015-06-09
