
连续性纯合片段在畜禽基因组研究中的应用
2019-04-22 11:54刘刚孙飞舟朱芳贤冯海永韩旭
遗传 2019年4期
刘刚,孙飞舟,朱芳贤,冯海永,韩旭
连续性纯合片段在畜禽基因组研究中的应用
刘刚,孙飞舟,朱芳贤,冯海永,韩旭
全国畜牧总站,北京 100193
随着高通量SNP芯片技术的快速发展和测序成本的大幅降低,SNP基因芯片和基因组重测序等技术被广泛地应用于畜禽基因组研究中。在基因组某一段区域内,当一定数量和一定密度的SNPs表现为纯合时,可以判定该区域存在连续性纯合片段(runs of homozygosity, ROH)。目前,连续性纯合片段已经逐渐成为分析畜禽群体近交程度、遗传结构等方面的重要指标之一。但是,ROH计算应用的评价标准还相对匮乏。本文系统介绍了连续性纯合片段的发展历史、原理、鉴定方法以及在畜禽群体结构解析、基因组功能分析和种畜禽品质检测等方面的应用情况,以期为畜禽遗传资源保种区和保种场在遗传多样性等动态监测方面提供参考。
高通量测序技术;连续性纯合片段;群体结构;基因组功能;遗传缺陷
单核苷酸多态性(single nucleotide ploymorhphisms, SNPs)是畜禽基因组中最常见的遗传变异,一般指在畜禽群体中频率大于1%单个核苷酸的变异,包括转换、颠换、缺失和插入。在基因组某一段区域内,当一定数量一定密度的SNPs表现为纯合时,可以判定该区域存在连续性纯合片段(runs of homozygosity, ROH)[1]。大量研究表明,ROH信息在畜禽、植物和人类群体近交程度和监测方面发挥着越来越重要的作用[2~6]。通过鉴别和分析ROH分布和频率等指标,可以深入剖析群体在世代间演变的历程,从而揭示这些群体经过系列变化后基因组中纯合片段的模式[7~9],也可以评估群体近交水平和群体中个体间的亲缘关系,进一步分析群体选择压力和交配模式等[10~12]。利用SNP基因芯片技术分析基因组中ROH是分析同源遗传关系(identical by descent, IBD)的有效方法[1,13]。通过SNP基因芯片技术对畜禽群体进行分析,可以获得同一群体不同世代动态变化的信息,如监测群体有效含量[14,15]和种公畜间近交系数[16]等。
1999年,Broman和Weber[7]首次发现并分析了人类染色体上长纯合片段,结果表明纯合片段的长短与人类健康相关。Gibson等[1]首次利用高密度SNP基因芯片技术分析了人类染色体上纯合片段的长度、频率和分布情况等,解析了人类基因组中存在ROH的机理[2,15,17]。随着畜禽SNP基因芯片和重测序技术的广泛应用[18~20],基于畜禽基因组信息的ROH研究也与日俱增。如Marras等[21]对牛()基因组ROH频率和分布情况等指标进行了分析;此外,在牛[4,10,21~24]、猪()[8,9,25~29]、马([30~33]、绵羊()[34~37]、山羊()[38,39]和鸡()[40~42]等畜禽群体结构和群体演变历史等研究中也利用了ROH特征信息。
本文主要综述了ROH的原理和方法以及在畜禽群体结构、基因组功能分析和种畜禽品质检测等方面的应用,以期为相关研究提供参考。
1 连续性纯合片段产生的原理
祖先单倍型相同的两个拷贝聚集在一个个体时会产生纯合片段,长单倍型片段来源于最近共同祖先,短单倍型片段来源于亲缘关系较远的共同祖先。由于亲缘关系较远个体基因组位点之间的强连锁不平衡形成了短ROH,不同类型群体可以产生长短不一的ROH发散分布。在远交群体中ROH产生取决于群体有效含量(),在小的群体中存在更多的ROH,而越大的群体会产生较少的ROH。由于混合种群血统来源于两个或者更多亲缘关系较远的群体,因而比它们祖先群体的ROH少。由于近交群体经历了瓶颈效应,尤其在个体间亲缘关系比较远的情况下,会产生数量较多的短ROH。但是,随着世代的交替,群体有效含量会越来越小,同时,由于近期近交事件的发生,从而在群体基因组片段中出现越来越多长短不一的ROH[43]。研究表明,ROH更多地富集在有害突变个体中,而在非有害突变个体中聚集较少;即使有害突变频率低于非有害突变的频率,ROH区域可能是有害突变个体发生突变的重要载体[16]。
2 检测连续性纯合片段的方法
根据不同类型数据的特点,可以制定适合于分析ROH的算法。目前分析方法主要包括观测基因型计数法和基于模型的分析方法。
2.1 观测基因型计数法
基因型计数法是根据设定杂合子最大数量和允许缺失基因型的数量,在基因组上鉴定连续纯合基因型的长片段。常用的软件有PLINK[44]、GERMLINE[45]和cgaTOH[46]等。Howrigan等[47]通过计算机模拟试验检测了已知120 Mb人基因组中纯合片段情况,其模拟结果表明,PLINK软件检测个体同一性的性能要优于GERMLINE软件。
2.2 基于模型的分析方法
基于模型的分析方法主要利用隐马尔可夫模型,分辨纯合子和杂合子基因组区域,获得等位基因频率和重组率等参数。常用的软件有BEAGLE[48]、H3M2[49]、FILTUS[50]、BCFtools/RoH[51]和GARLIC[52]等。全基因组重测序深度的增加有利于减少基因型判定的错误率,从而改进隐马尔可夫模型的判定,大大提高检测ROH的精度,进一步确定较短ROH在近交衰退中的作用。
目前,PLINK软件广泛应用于ROH分析中。不同畜禽群体中鉴定ROH不同软件设置参数详见表1。
3 连续性纯合片段信息在畜禽基因组研究中的应用
3.1 亲缘关系的鉴定
随着高通量测序技术的迅猛发展,利用基因组信息分析个体和群体间的近交程度越来越被关注,尤其是检测染色体上ROH的长度和分布情况,从而间接地分析群体中个体间的近交程度。通过计算基因组中特定长度(如>1 Mb、>2 Mb、>4 Mb、>8 Mb、>16 Mb等)ROH的值反映群体在基因组水平上的近交程度。
近交系数传统分析方法是假定群体中祖先没有亲缘关系的前提下,通过通径原理分析计算得到的。目前,随着高密度SNP基因芯片技术的广泛应用,在不同畜禽群体中利用基因组信息分析真实的基因组近交程度成为可能。研究表明,基因组信息估测近交程度比传统意义上的系谱信息更有效[4,10,79]。利用系谱信息估测亲缘关系是通过基因组IBD概率的统计期望值,而利用基因组信息估测的是个体间实际的亲缘关系[80,81]。不同畜禽群体中鉴定ROH信息以及基于系谱信息和基因组信息近交系数的相关系数见表2。
表1 不同畜禽群体中鉴定ROH设置参数比较
Table 1 Comparison of pre-set parameters for identification and characterization of ROH in different animal species
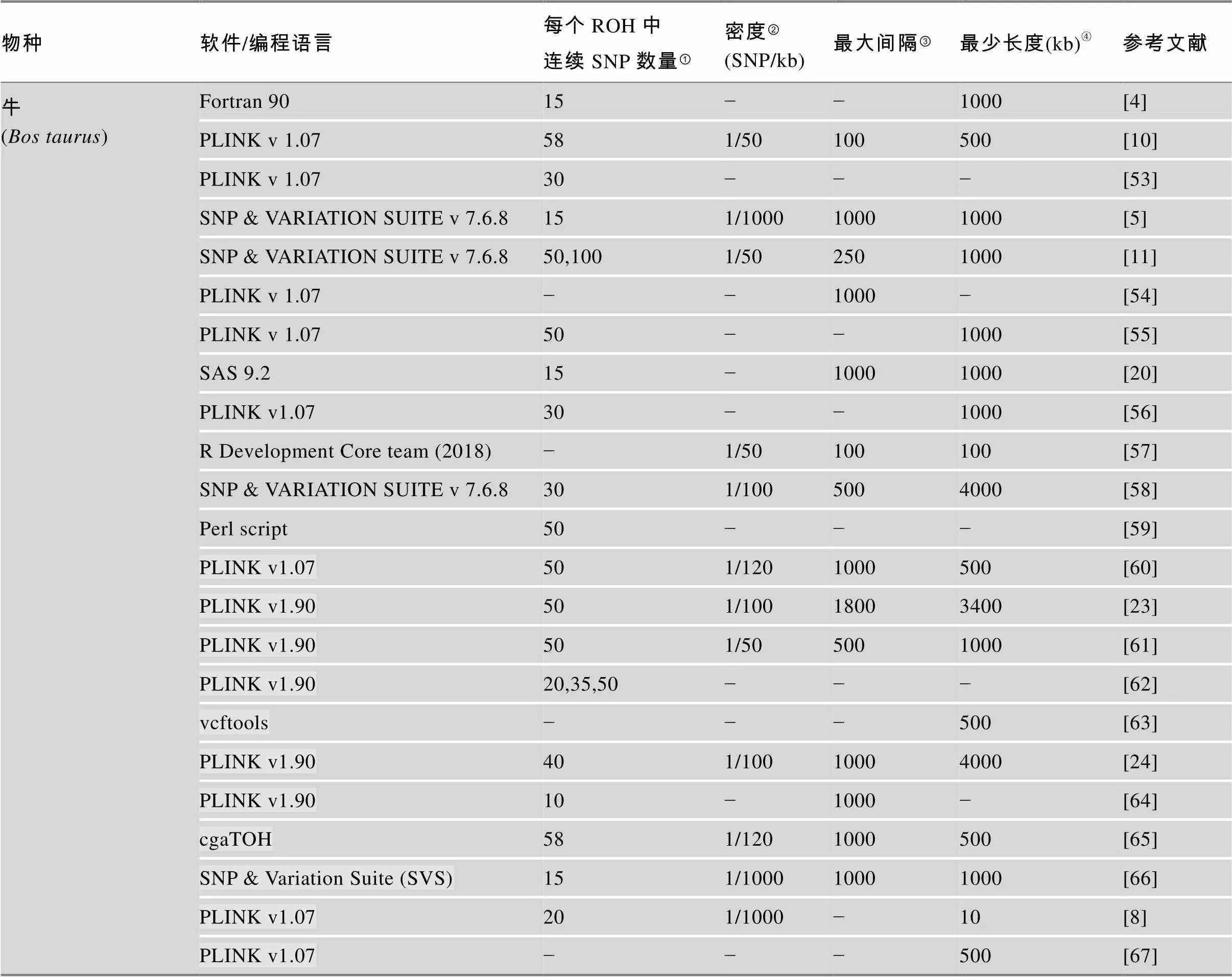
物种软件/编程语言每个ROH中连续SNP数量①密度②(SNP/kb)最大间隔③最少长度(kb)④参考文献 牛 (Bos taurus)Fortran 9015−−1000[4] PLINK v 1.07581/50100500[10] PLINK v 1.0730−−−[53] SNP & VARIATION SUITE v 7.6.8151/100010001000[5] SNP & VARIATION SUITE v 7.6.850,1001/502501000[11] PLINK v 1.07−−1000−[54] PLINK v 1.0750−−1000[55] SAS 9.215−10001000[20] PLINK v1.0730−−1000[56] R Development Core team (2018)−1/50100100[57] SNP & VARIATION SUITE v 7.6.8301/1005004000[58] Perl script50−−−[59] PLINK v1.07501/1201000500[60] PLINK v1.90501/10018003400[23] PLINK v1.90501/505001000[61] PLINK v1.9020,35,50−−−[62] vcftools−−−500[63] PLINK v1.90401/10010004000[24] PLINK v1.9010−1000−[64] cgaTOH581/1201000500[65] SNP & Variation Suite (SVS)151/100010001000[66] PLINK v1.07201/1000−10[8] PLINK v1.07−−−500[67]
续表
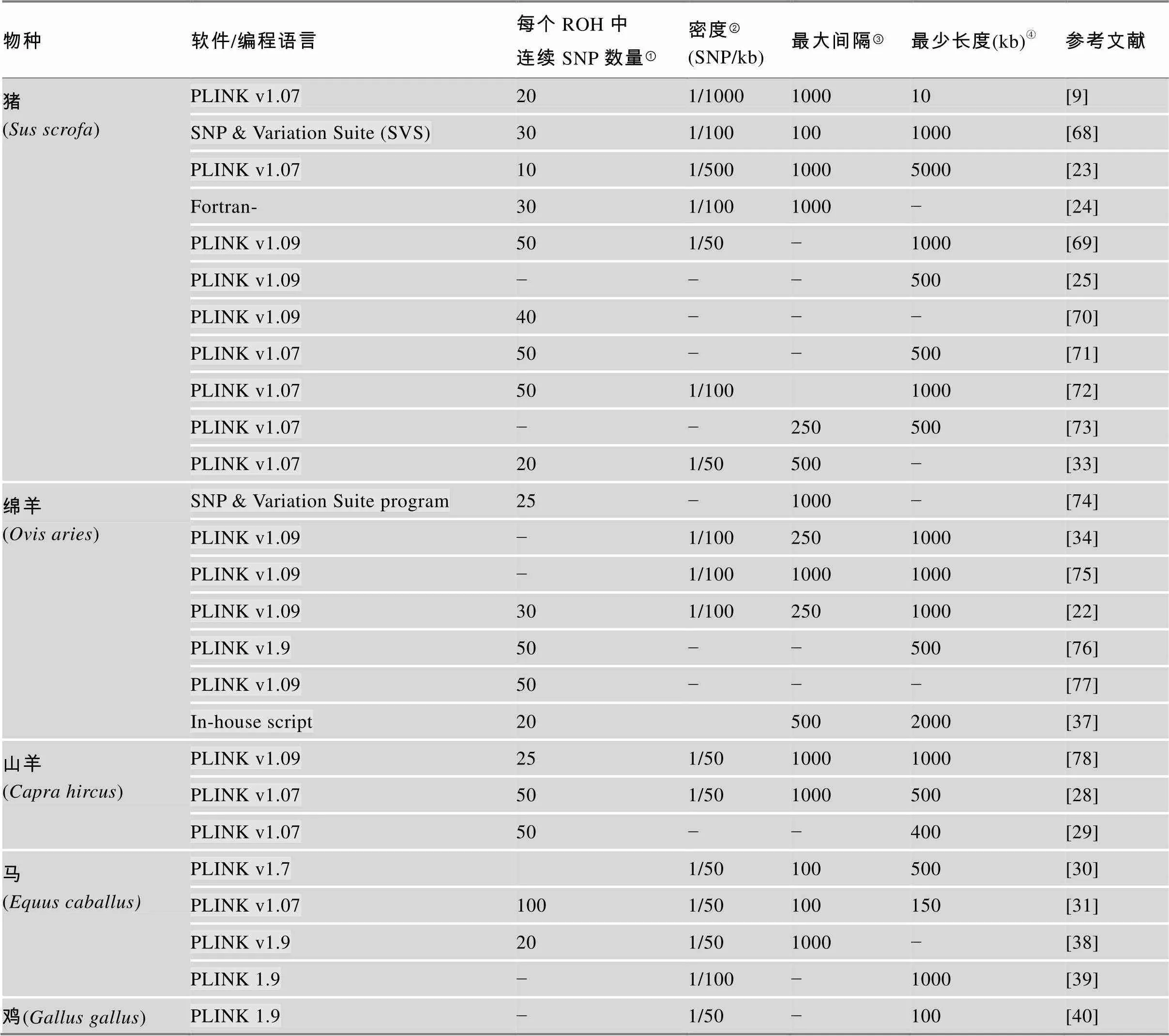
物种软件/编程语言每个ROH中连续SNP数量①密度②(SNP/kb)最大间隔③最少长度(kb)④参考文献 猪 (Sus scrofa)PLINK v1.07201/1000100010[9] SNP & Variation Suite (SVS)301/1001001000[68] PLINK v1.07101/50010005000[23] Fortran-301/1001000−[24] PLINK v1.09501/50−1000[69] PLINK v1.09−−−500[25] PLINK v1.0940−−−[70] PLINK v1.0750−−500[71] PLINK v1.07501/1001000[72] PLINK v1.07−−250500[73] PLINK v1.07201/50500−[33] 绵羊 (Ovis aries)SNP & Variation Suite program25−1000−[74] PLINK v1.09−1/1002501000[34] PLINK v1.09−1/10010001000[75] PLINK v1.09301/1002501000[22] PLINK v1.950−−500[76] PLINK v1.0950−−−[77] In-house script205002000[37] 山羊 (Capra hircus)PLINK v1.09251/5010001000[78] PLINK v1.07501/501000500[28] PLINK v1.0750−−400[29] 马 (Equus caballus)PLINK v1.71/50100500[30] PLINK v1.071001/50100150[31] PLINK v1.9201/501000−[38] PLINK 1.9−1/100−1000[39] 鸡(Gallus gallus)PLINK 1.9−1/50−100[40]
①表示一个ROH片段中连续SNP位点数量;②表示在每个运行单元中SNPs的密度;③表示连续纯合子片段之间的最大间隔;④表示鉴定ROH的最小长度。“−”表示无此信息。
表2 畜禽群体中鉴定ROH信息以及基于系谱信息和基因组信息近交系数的相关系数统计表
Table 2 Studies of ROH and correlations between the inbreeding from pedigree data and from genome data through ROH in livestock and poultry species

物种品种/群体数量ROH平均数量ROH平均长度(Mb)相关系数参考文献 FPED①, FROHFPED, FROH>1 MbFPED, FROH>2 MbFPED, FROH>4 MbFPED, FROH>8 MbFPED, FROH>16 Mb 牛 (Bos taurus)Austrian Simmental500−−−0.640.670.680.680.63[4] Multiple breeds891−0.30~5.09②0.71−−−−−[10] Brown Swiss30498.91.30−0.660.67−0.600.50[5] Fleckvieh50294.50.440.660.690.700.64 Nowegian Red49880.00.510.610.610.620.53 Tyrol Grey11772.31.880.710.720.710.70
续表

物种品种/群体数量ROH平均数量ROH平均长度(Mb)相关系数参考文献 FPED①, FROHFPED, FROH>1 MbFPED, FROH>2 MbFPED, FROH>4 MbFPED, FROH>8 MbFPED, FROH>16 Mb 牛 (Bos taurus)Italian Holstein209381.73.6−0.70−0.690.650.56[20] Italian Brown74994.63.90.660.660.650.58 Italian Simmental47994.32.20.660.740.760.71 Jersey1602−−0.70③/0.71④−−−−−[57] Cinisara719.3813.570.45−−−−−[82] Modicana72110.312.310.27 Reggiana16810.4210.160.31 Italian Holstein967.1511.780.44 Holstein210721.28.020.73−−−−−[83] Maasai−10317.460.90−−−−−[84] Tarime5613.120.75 Sukuma3610.650.61 Boran999.480.56 Friesian1559.680.54 Brown Swiss28121.022640.45−−−−−[23] Braunvieh338618.6184.6 Origianl Braunvieh1678.473.7 Holstein256814.2145.2 Red Holstein196011.2112.1 Swiss Fleckvieh5477.175.6 Simmental24810.996.6 Eringer368.566.2 Evolèner2115.5185.7 猪 (Sus scrofa)Iberian64−−−0.77−−0.81−[68] Yorkshire2358−−0.69−−−−−[23] Guadyerbas109−−0.63-0.24−−0.60−[24] Landrace117852.7252.90.24−−−−−[69] Large White120061.4280.10.015 Duroc106616.726.750.31−−−−−[70] Landrace76823.1911.270.32 Yrokshire111125.8811.990.53 Crossbred1128.252.60.00 马 (Equus caballus)Sorraia2⑤4175⑥0.19−−−−−−[29] Dülmen Horse1⑤2804⑥0.14 Arabian1⑤3581⑥0.15 Saxon-Thuringian1⑤3138⑥0.15 Thoroughbred1⑤4595⑥0.20 Hanoverian4⑤311⑥0.14
续表
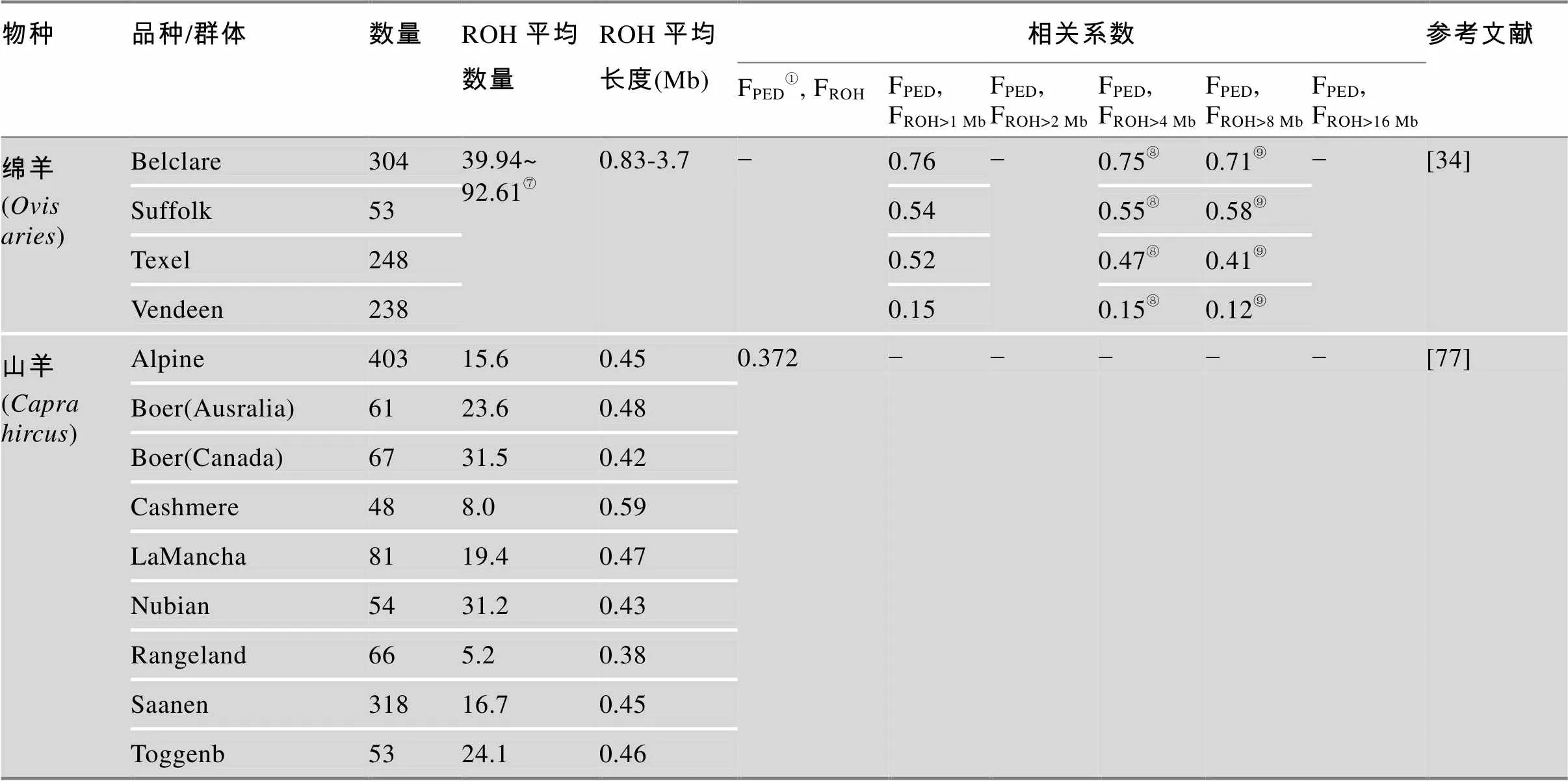
物种品种/群体数量ROH平均数量ROH平均长度(Mb)相关系数参考文献 FPED①, FROHFPED, FROH>1 MbFPED, FROH>2 MbFPED, FROH>4 MbFPED, FROH>8 MbFPED, FROH>16 Mb 绵羊 (Ovis aries)Belclare30439.94~ 92.61⑦0.83-3.7−0.76−0.75⑧0.71⑨−[34] Suffolk530.540.55⑧0.58⑨ Texel2480.520.47⑧0.41⑨ Vendeen2380.150.15⑧0.12⑨ 山羊 (Capra hircus)Alpine40315.60.450.372−−−−−[77] Boer(Ausralia)6123.60.48 Boer(Canada)6731.50.42 Cashmere488.00.59 LaMancha8119.40.47 Nubian5431.20.43 Rangeland665.20.38 Saanen31816.70.45 Toggenb5324.10.46
①根据系谱信息计算的近交系数;②估计平均ROH长度为ROH平均覆盖基因组长度与ROH总数量的平均值;③以连续100个纯合子SNPs鉴定为一个ROH;④以连续30、50、80个纯合子SNPs鉴定为一个ROH;⑤序列信息从NCBI获得;⑥使用50 SNPs滑动窗口定义的值;⑦品种间变化范围为39.94~92.61 Mb,每个品种平均ROH变化范围为0.83~3.7 Mb(ROH≥20 Mb);⑧基因组中5 Mb计算的FROH;⑨基因组中10 Mb计算的FROH。“−”表示无此信息。
目前,基于基因组信息估测近交程度的方法主要有以下3种:(1)基于ROH的近交系数(FROH),是指ROH片段长度之和占整个基因组总长度的比例。McQuillan等[2]引入FROH作为检测个体间同一性指标,其中计算公式中整个基因组是指基因组常染色体上特定区域的长度,不同的研究中设置的具体参数不同;(2)标记基因型中纯合子所占的比例(FHOM),即所检测SNP中的纯合子比例;(3)基于基因组关系矩阵的近交系数(FGRM),其中G矩阵计算方法参考文献[85]。杨湛澄等[83]利用高密度SNP 标记通过两种基因组近交计算方法(FROH和FHOM)分析中国荷斯坦牛基因组近交程度,其结果表明,共检测到44 676个ROH片段,ROH在染色体上并非均匀分布,其长度主要分布在1~10 Mb之间。两种基因组近交系数之间的相关性比较大,而基因组近交系数与系谱近交之间的相关性较低。Peripolli等[61]采用4种近交系数计算方法(FPED、FHOM、FGRM和FROH)对瘤牛群体近交程度进行了评估,结果表明,FROH和FGRM相关性为弱到中度相关;FROH和FHOM相关性从弱到强相关;FPED和FHOM与FGRM和FHOM之间的相关程度为中等;FROH和FPED相关系数随着ROH长度的增加而增大。因此,在群体系谱信息缺失的情况下,FROH可以作为替代方法评价畜禽群体的近交程度。
Keller等[6]研究表明,FROH指标与FPED指标相比,具有以下几方面优点:(1) FROH可以更准确估计共同祖先甚至50代前后代个体基因组中纯合性状态;(2)在系谱信息不完整或者缺失的情况下,FROH指标可以检测基因组中纯合片段分布,同时可以发现与纯合性高的特异性位点;(3) FPED指标是相对于基础群而言的,在基础群假定祖先个体的基因组没有选择和重组事件的发生。此外,减数分裂是一个随机过程,子代获得父母双方遗传物质的过程存在着随机变异,且这样的变异随着减数分裂的增加而增加,而FPED仅是IBD概率的期望值。从表2统计结果看出,在牛和猪品种鉴定ROH研究中,FROH和FPED之间的相关程度为中度或者高度,因此可以仅采用FROH监测牛和猪群体的近交程度。也有研究表明,鉴定ROH的长度与FROH和FPED之间相关程度为正相关(表2),ROH反映了群体过去和现在的亲缘关系,而FPED仅根据现有的系谱记录数据估测近交程度。随着群体系谱信息的不断积累,基于系谱近交系数与基于基因组近交系数的相关性也随之增加[20]。根据Saura等[26]报道,当ROH长度大于5 Mb时,计算的FROH值和FPED值接近,而当ROH长度小于5 Mb时,计算的FROH值比FPED值小4倍多。利用FROH和FPED两种方法估测了和牛群体中个体亲缘关系,其结果表明采用系谱信息数据低估了和牛群体的近交程度,基因组近交系数可以反映真实的近交程度,该结果与已有的研究结果一致[20,24,57]。Metzger等[31]估测了马基因组近交系数,在一个窗口滑动50个SNP条件设置下,FROH值变化范围为0.18~ 0.43。Guangul等[38]估测了5个山羊群体的基因组近交程度,ROH长度从1~16 Mb,其FROH的值从0.0500~0.0048。Brito等[77]采用50K基因芯片通过4种不同的近交系数对9个山羊群体近交程度进行了评估,其中基于系谱和ROH近交系数的相关系数为0.372;基于基因型计数方法和ROH近交系数的相关性高达0.901;而基于ROH和基于VanRaden与基于Leuenegger方法的近交系数均为负相关(相关系数分别为-0.133和-0.264)。Grossi等[70]分析了杜洛克、长白和大约克夏纯种猪以及长白大约克夏猪杂交F1代4个群体共计3057个个体ROH分布情况,其结果表明每个个体ROH平均长度在4个群体中依次为16.72、23.19、25.88和8.25;平均数量分别为6.75 Mb、11.28 Mb、11.99 Mb和2.65 Mb。FPED和FROH相关系数在4个群体中依次为0.31、0.32、0.53和0.00;FEH和FROH相关系数依次为0.41、0.72、0.69和0.64 (表2)。Kim等[63]采用重测序技术对经过选育的126头Hanwoo牛个体进行了检测,通过遗传改良提高了其群体的体重,但是群体的近交程度有所增加,其FROH值比未改良的群体升高了约0.02。通过4种类型近交系数评估瘤牛群体的近交程度,其研究结果表明FPED的值变化范围为0.00~0.327;FROH值变化范围为0.001~0.201。FPED与FROH相关系数和FGRM与FROH相关系数从弱相关变为中等相关,其变化范围从-0.11~0.51;FROH和FROM相关系数从弱相关到强相关;不同长度估测的FROH和FPED相关系数随着ROH长度的增加而增加[61]。通过ROH方法对中国白耳黄鸡、北京油鸡和狼山鸡3个群体的保种效果进行评估,检测到基于系谱的近交系数为0.0789 (白耳黄鸡)~0.2010 (北京油鸡);通过几个世代的保种效果监测,表明其基于系谱的近交系数在其群体中变动幅度比较小,而检测到基于ROH近交系数的值要比基于系谱的值要偏低,其值为0.0511 (白耳黄鸡)~0.0745 (北京油鸡),基于系谱和基于ROH近交系数的相关系数为0.76[25]。综上所述,评估畜禽群体的近交程度,FROH是比较有效的评价指标,可以很好地补充由于系谱信息预测群体近交程度的不足,也可以通过鉴定ROH片段提高IBD片段定位的精度。
3.2 近交衰退的评估
Garrod等[86]发现一些人类疾病,如白血病、尿黑酸尿等,这些遗传疾病在近亲婚姻后代个体中发病率比较高,尤其在近交个体的隐性携带者,通过长纯合子片段可以检测到致病的隐性有害变异。Zhang等[25]发现有害纯合变异体和基因组中ROH片段之间呈现线性相关,致病座位有害基因纯合子出现在ROH上的频率要高于正常基因的频率。Szpiech等[87]研究结果表明,鉴定的ROH高覆盖度片段中包含有较长有害变异区段,这也与引起近交衰退有害基因变异位点一般以纯合子状态存在假设是一致的。Muchadeyi等[35]在南非洲波斯羊3、4和25号染色体上检测到ROH片段上与神经系统、骨骼和大脑发育相关的基因,如基因、基因和基因。Huson等[88]利用基因组关联分析,结合单倍型分析、选择信号分析和ROH分析共同鉴定了牛20号染色体上位点。Mészáros等[56]采用ROH和基因组关联分析发现了弗莱维赫牛眼脸内翻遗传缺陷基因组区段。Pryce等[89]基于系谱信息的近交系数估测了奶牛产量和个体健康性状,其研究结果表明群体中近交程度增加1%,一个哺乳期内荷斯坦牛和泽西奶牛奶产量分别减少21 L和12 L。Kim等[63]分析了近50年来美国泽西牛基因组中增加的60多个ROH区域与系谱信息估测的近交增量呈正相关,在3号、7号、8号和12号染色体上鉴定的ROH与后代女儿繁殖率呈负相关,体细胞评分的结果与繁殖性状的结果相似。由于近交衰退引起1号、3号、4号、5号和13号染色体上增加的ROH影响了体细胞评分的结果,染色体上高度纯合性导致繁殖率的下降和乳房炎易感性的增加。Silió等[68]研究了近交衰退对断奶后仔猪生产性能的影响,结果表明由于群体近交系数增加,导致其断奶仔猪生产性能下降,具体表现为近交系数每增加0.1,其日增重减少4.4%,90日龄体重减少1.52%。Saura等[26]分析了伊比利亚猪两个高度近交系中的繁殖性状,近交系数每增加0.1,其仔猪初生后存活率和仔猪出生后总数量有下降的趋势。Ferenčaković等[5]研究牛群体中ROH分布情况,解析了在群体近交增量增加情况下牛精液品质下降的机理,发现与精子数量相关ROH区域有4个,与精子活力相关ROH区域有5个,但是同时与精子数量和精子活力相关ROH区域仅为1个。
3.3 遗传多样性分析
获得大量畜禽基因组信息使得人们更好地分析畜禽群体遗传多样性等指标。维持群体遗传多样性是畜禽保种的重要任务之一,以便利用更丰富的育种素材获得动物产品。采用基因组信息分析共祖先策略已经应用于保护群体遗传多样性和近交增量的分析中[90]。当保种群体中出现中高近交繁殖的迹象时,基于IBD方法分析共同祖先可以作为一个策略维持遗传多样性和保种计划的适合度[91]。因此,较小的群体有效含量和较高的近交增量会降低群体遗传多样性,通过畜禽保种方案的有效实施,监测群体的遗传变异,防止群体中发生不可逆转遗传多样性的减少,最大限度地增加保种群体适应外部环境变化的能力。Fleming等[40]采用600K基因芯片分析了非洲3个鸡群体的遗传多样性,结果表明,群体中所有染色体仅有16号染色体上没有检测到ROH,每个个体ROH在基因组的覆盖程度为2%~40%。Mastrangelo等[24]为了更好地制定和实施保种计划,分析了30个意大利牛群体遗传多样性,结果表明观测杂合度的值变化范围为0.297~0.358,期望杂合度的值变化范围为0.267~0.353。在祖先群体中群体有效含量较高,但是Pontremolese和Mucca Pisana2个群体有效含量比较低。通过分析个体ROH分布和长度等参数有助于畜禽保种项目的制定和实施,在Pontremolese、Varzese-Ottonese和Mucca Pisana群体中检测到高水平的ROH,如尤其针对这些群体,在实施配种计划中尽量增加种公畜血统,减少其遗传多样性的损失,维持或者增加其群体有效含量。Zhang等[42]采用ROH方法对中国白耳黄鸡、北京油鸡和狼山鸡3个保种群体的遗传多样性、基因组近交系数和纯合性进行分析,经过实施近10年的保种策略,白耳黄鸡和北京油鸡群体的遗传多样性有所下降,狼山鸡群体的遗传多样性有上升的趋势。
3.4 人工选择的追踪
基因组中鉴定的选择信号揭示了驯化群体中双向选择的痕迹。与没有受到人工选择的群体比较,对于优秀种畜禽个体的选育,降低了其群体表型的多样性和重塑了基因组,其中包括基因组中ROH存在的模式[12]。有研究表明,对于选育的优秀种畜禽个体使其基因组中单倍型多样性下降,同时也增加了选择位点相邻位点的纯合性,导致其受到选择区域中的ROH频率增加[11]。ROH并不是随机分布在基因组中,大部分ROH出现在受选择区域。基因组中受选择的区域倾向于产生“ROH岛”,相对于基因组其他区域,这些区域遗传多样性低,纯合性比较高。Purfield等[10]研究了牛基因组中出现ROH频率较高的4条染色体,其中在ROH区域中包含了影响牛免疫力、胴体和难产等重要性状的主效基因。在不同的阿拉伯马群体中也开展了ROH的研究,分析了受到正向选择区域的ROH。Metzger等[31]研究了马基因组中受到选择和未受到选择区域中ROH的功能分布,发现了与细胞代谢、生长发育和免疫系统相关的候选基因。Fleming等[41]采用FST、综合单倍型评分(integrated haplotype score)和ROH等信息检测了在非洲和北非不同生态环境中生长鸡品种的选择信号,分析表明非洲生长的鸡群体选择倾向于热应激和血管生成,而北非群体更倾向于能量平衡,其中鸡品种基因组中2号和3号染色体在不同群体中差异最大。通过长期优秀种畜的选育,群体选择强度增加和有效群体含量减少有可能会导致群体生存力和多样性受到威胁。在畜禽选育和保种过程中,尽量避免群体遗传变异性减少,避免基因组中有害基因的表达。人工选择会导致群体近交系数的增加,因此要采取有效的措施控制近交程度的增加。另外,随着人工授精技术的应用,用于采精的优秀种公牛近交程度也影响着整个配种群体的近交程度[63]。
3.5 功能基因的筛选
Bosse等[8]利用重测序技术和SNP基因芯片技术检测了猪基因组上纯合区域,在欧洲猪品种中发现两个重叠ROH区域,该区域上有与神经系统发育细胞分化相关的11个基因,这些基因在大白猪和利比里亚猪中被验证表达存在差异。在亚洲品种中存在4个共享区域,其中有一个重叠区域仅存在亚洲野猪中,该区域中包括91个基因,并且已经有相关报道表明该区域在亚洲猪品种中经过了正向选择;其中在5号染色体上另一个共享区域包括与氧化还原反应相关的和基因,与脂肪细胞分化正向调控的基因。在非洲3个鸡品种中一致的ROH区域内比对发现与脂肪代谢、免疫功能和热激介导相关的基因(FDR<0.15),选择区域内也发现与健康和氧化应激反应相关的基因[38]。通过瘤牛群体中ROH分析,发现群体基因组中有7.01% (175.28 Mb)为纯合区域,在整个群体中鉴定的ROH 14个区域的频率高于50%,发现与泌乳()、产奶量和乳成分(和)、热适应(、和)等相关候选基因[61]。Metzger等[31]采用全基因组测序方法分析了英国设得兰群岛上2个微型矮马群体和1个正常体高矮马群体,发现在这2个微型矮马群体和1个正常体高矮马群体中ROH区域内存在4个变异,这4个变异解释了设得兰群岛上矮马群体和其他正常体高马群体中72%体高变异效应。
3.6 种畜禽品质检测
在瑞士Appenzeller Barthuhn鸡群体中存在一种十字鸡喙的遗传缺陷,Joller等[92]在该群体和正常群体中通过检测基因组ROH对存在十字鸡喙个体的遗传机理进行研究,初步假定角蛋白家族基因为十字鸡喙遗传缺陷的候选基因,在编码区内发现有两个显著的同义突变,但是十字鸡喙遗传缺陷的遗传机理还有待于进一步研究确认。目前,利用ROH检测种畜禽品质的报道还比较少。通过基因组中ROH信息剖析畜禽遗传缺陷的机制,明确致病基因,采用快速有效的方法进行检测,进一步规范种畜禽市场。我国是畜禽资源大国,据不完全统计,截止2018年12月,我国地方畜禽遗传资源数量为556个,国家级保护区数量为24个,国家级保种场数量为165个。如何利用应用成熟的现代生物技术手段对我国畜禽遗传资源群体进行动态监测,尤其是国家级保种场畜禽群体的动态变化情况,已经成为当前畜禽遗传资源保护领域亟待解决的问题。目前,ROH在不同畜禽基因组中的广泛应用为解决这一难题提供了一定的措施。对于群体动态监测而言,主要监测群体近交程度、遗传多样性、群体结构以及种群特性生产性状等变化情况等。近交系数最初由Wright S. (1921年)提出,在假定群体中祖先没有亲缘关系的前提下,通过通径原理分析计算得到的。利用系谱信息估测亲缘关系是通过基因组IBD概率的统计期望值,而利用基因组信息可以估测个体间实际的亲缘关系。在系谱信息缺少的情况下,可以采用FROH估计其群体近交系数。如果ROH>5 Mb时,其基于系谱估测的平均值与FROH值相关系数为0.87,而当ROH<5 Mb时,其基于系谱估测平均值与FROH值相关性较小[16,24],在实际应用中,可以结合系谱信息,采用较大的ROH估测群体基因组近交系数。近交群体会产生近交衰退现象,近交衰退是由于基因组纯合片段增多引起的现象,在生产实践中,由于近交衰退导致群体整体生产性能会逐渐下降,对于畜禽保种和育种管理者而言,研究近交衰退以及由此引起群体生产性能下降是一个比较重要的课题。采用ROH信息已经成功定位人类许多罕见隐性疾病的致病基因[41],这对于研究群体种公畜遗传缺陷的致病机理具有很高的借鉴作用,也为规范种畜禽市场提供检测依据。另外,充分利用ROH信息挖掘畜禽群体适应性、繁殖力、耐粗饲等性状的特有基因更有利于畜禽保种场保护与利用工作的有序开展。在生物大数据时代下,畜禽遗传资源保护与利用工作也需要不断调整研究思路和策略来迎合和充分利用高通量测序技术进步带来的福祉。
4 结语与展望
本文全面总结了畜禽基因组中ROH发展历史、鉴定方法以及在群体结构、基因组功能分析和种畜禽品质检测等方面的应用。综上所述,ROH在畜禽基因组中是普遍存在的,通过分析基因组中分布的ROH,人们可以了解群体近交程度、群体多样性以及种公畜(禽)遗传缺陷等。但是,目前研究的物种主要集中在奶牛和猪中,在肉牛和其他家畜以及家禽中研究的较少,今后需要加大对马、驴、绵羊、山羊和家禽等畜禽基因组中ROH的研究,从而更好地了解ROH在染色体上分布情况以及其作用机理。
目前,鉴定畜禽基因组中ROH没有统一的标准,在不同畜种的研究中采用不同算法和方法。迄今为止,已有的研究很少关注优化鉴定ROH的参数组合,如果使用最优参数组合会更好地理解基因组中纯合性形成的机制[81]。此外,畜禽基因组中鉴定ROH频率和分布受到许多因素的影响,ROH在染色体内和染色体之间分布频率差异大,因此在染色体上会出现ROH集中区域(也称ROH岛),也会出现ROH分布少的区域(也称ROH荒漠),但相关机理还有待于进一步研究。
2015年,动物基因组功能注解(Functional Annotation of Animal Genomes, FAANG)计划启动,充分说明农业动物领域相关研究的重要性[93]。随着畜禽基因组研究时代的到来,海量数据的获得便于更加系统地研究ROH特征序列、进一步剖析群体近交增量、群体演变历史、选择信号以及遗传疾病等机理,从而开启畜禽基因组研究运用于畜禽遗传资源保护与利用的新时代[94]。
[1] Gibson J, Morton NE, Collins A. Extended tracts of homozygosity in outbred human populations., 2006, 15(5): 789–795.
[2] McQuillan R, Leutenegger AL, Abdel-Rahman R, Franklin CS, Pericic M, Barac-Lauc L, Smolej-Narancic N, Janicijevic B, Polasek O, Tenesa A, Macleod AK, Farrington SM, Rudan P, Hayward C, Vitart V, Rudan I, Wild SH, Dunlop MG, Wright AF, Campbell H, Wilson JF. Runs of homozygosity in European populations., 2008, 83(3): 359–372.
[3] Johnson EC, Evans LM, Keller MC. Relationships between estimated autozygosity and complex traits in the UK Biobank., 2018, 14(7): e1007556.
[4] Ferenčaković M, Hamzic E, Gredler B, Curik I, Sölkner J. Runs of homozygosity reveal genome-wide autozygosity in the Austrian Fleckvieh cattle., 2011, 76: 286–293.
[5] Ferenčaković M, Hamzić E, Gredler B, Solberg TR, Klemetsdal G, Curik I, Sölkner J. Estimates of autozygosity derived from runs of homozygosity: empirical evidence from selected cattle populations., 2013, 130(4): 286–293.
[6] Keller MC, Visscher PM, Goddard ME. Quantification of inbreeding due to distant ancestors and its detection using dense single nucleotide polymorphism data., 2011, 189(1): 237–249.
[7] Broman KW, Weber JL. Long homozygous chromosomal segments in reference families from the centre d'Etude du polymorphisme humain., 1999, 65(6): 1493–1500.
[8] Bosse M, Megens HJ, Madsen O, Paudel Y, Frantz LA, Schook LB, Crooijmans RP, Groenen MA. Regions of homozygosity in the porcine genome: consequence of demography and the recombination landscape., 2012, 8(11): e1003100.
[9] Herrero-Medrano JM, Megens HJ, Groenen MA, Ramis G, Bosse M, Pérez-Enciso M, Crooijmans RP. Conservation genomic analysis of domestic and wild pig populations from the Iberian Peninsula., 2013, 14: 106.
[10] Purfield DC, Berry DP, McParland S, Bradley DG. Runs of homozygosity and population history in cattle., 2012, 13: 70.
[11] Kim ES, Cole JB, Huson H, Wiggans GR, Van Tassell CP, Crooker BA, Liu G, Da Y, Sonstegard TS. Effect of artificial selection on runs of homozygosity in u.s. Holstein cattle., 2013, 8(11): e80813.
[12] Zhang Q, Guldbrandtsen B, Bosse M, Lund MS, Sahana G. Runs of homozygosity and distribution of functional variants in the cattle genome., 2015, 16: 542.
[13] Lencz T, Lambert C, DeRosse P, Burdick KE, Morgan TV, Kane JM, Kucherlapati R, Malhotra AK. Runs of homozygosity reveal highly penetrant recessive loci in schizophrenia., 2007, 104(50): 19942–19947.
[14] Megens HJ, Crooijmans RP, Bastiaansen JW, Kerstens HH, Coster A, Jalving R, Vereijken A, Silva P, Muir WM, Cheng HH, Hanotte O, Groenen MA. Comparison of linkage disequilibrium and haplotype diversity on macro- and microchromosomes in chicken., 2009, 10: 86.
[15] Kirin M, McQuillan R, Franklin CS, Campbell H, McKeigue PM, Wilson JF. Genomic runs of homozygosity record population history and consanguinity., 2010, 5(11): e13996.
[16] Curik I, Ferenčaković M, Sölkner J. Inbreeding and runs of homozygosity: a possible solution to an old problem., 2014, 166(1): 26–34.
[17] Nothnagel M, Lu TT, Kayser M, Krawczak M. Genomic and geographic distribution of SNP-defined runs of homozygosity in Europeans., 2010, 19(15): 2927–2935.
[18] Song NN, Zhong JC, Chai ZX, Wang Q, He SM, Wu JB, Jian SL, Ran Q, Meng X, Hu HC. The whole genome data analysis of Sanjiang cattle., 2017, 50(01): 183–194.宋娜娜, 钟金城, 柴志欣, 汪琦, 何世明, 吴锦波, 蹇尚林, 冉强, 蒙欣, 胡红春. 三江黄牛全基因组数据分析. 中国农业科学, 2017, 50(1): 183–194.
[19] Lan R, Zhu L, Shao QY, Hong QH. Whole-genome resequencing in Yunnan black goat., 2016, (05): 11–17.兰蓉, 朱兰, 邵庆勇, 洪琼花. 云南黑山羊全基因组重测序. 草食家畜, 2016, (5): 11–17.
[20] Mei CG, Wang HC, Zan LS, Cheng G, Li AP, Zhao CP, Wang HB. Research progress on animal genome research based on high-throughput sequencing technology.,2016, 44(3): 43–51.梅楚刚, 王洪程, 昝林森, 成功, 李安宁, 赵春平, 王洪宝. 基于高通量测序的动物基因组研究进展. 西北农林科技大学学报(自然科学版), 2016, 44(3): 43–51.
[21] Marras G, Gaspa G, Sorbolini S, Dimauro C, Ajmone- Marsan P, Valentini A, Williams JL, Macciotta NP. Analysis of runs of homozygosity and their relationship with inbreeding in five cattle breeds farmed in Italy., 2015, 46(2): 110–121.
[22] Williams JL, Hall SJ, Del Corvo M, Ballingall KT, Colli L, Ajmone Marsan P, Biscarini F. Inbreeding and purging at the genomic Level: the Chillingham cattle reveal extensive, non-random SNP heterozygosity., 2016, 47(1): 19–27.
[23] Signer-Hasler H, Burren A, Neuditschko M, Frischknecht M, Garrick D, Stricker C, Gredler B, Bapst B, Flury C. Population structure and genomic inbreeding in nine Swiss dairy cattle populations., 2017, 49(1): 83.
[24] Mastrangelo S, Sardina MT, Tolone M, Di Gerlando R, Sutera AM, Fontanesi L, Portolano B. Genome-wide identification of runs of homozygosity islands and associated genes in local dairy cattle breeds., 2018, 1–9.
[25] Zhang Y, Young JM, Wang C, Sun X, Wolc A, Dekkers JCM. Inbreeding by pedigree and genomic markers in selection lines of pigs. In:Proceedings of the 10thWorld Congress of Genetics Applied to Livestock Production. Vancouver, BC, Canada, 2014.
[26] Saura M, Fernández A, Varona L, Fernández AI, de Cara MÁ, Barragán C, Villanueva B. Detecting inbreeding depression for reproductive traits in Iberian pigs using genome-wide data., 2015, 47: 1.
[27] Traspov A, Deng W, Kostyunina O, Ji J, Shatokhin K, Lugovoy S, Zinovieva N, Yang B, Huang L. Population structure and genome characterization of local pig breeds in Russia, Belorussia, Kazakhstan and Ukraine., 2016, 48: 16.
[28] Yang B, Cui L, Perez-Enciso M, Traspov A, Crooijmans RPMA, Zinovieva N, Schook LB, Archibald A, Gatphayak K, Knorr C, Triantafyllidis A, Alexandri P, Semiadi G, Hanotte O, Dias D, Dovč P, Uimari P, Iacolina L, Scandura M, Groenen MAM, Huang L, Megens HJ. Genome-wide SNP data unveils the globalization of domesticated pigs., 2017, 49(1): 71.
[29] Lago LV, Nery da Silva A, Zanella EL, Groke Marques M, Peixoto JO, da Silva MVGB, Ledur MC, Zanella R. Identification of genetic regions associated with scrotal hernias in a commercial swine herd., 2018, 5(1).doi:10.3390/vetsci5010015.
[30] Khanshour AM. Genetic diversity and population structure of the Arabian horse populations from Syria and other countries[D]. Texas A&M University, College Station, 2013a.
[31] Metzger J, Karwath M, Tonda R, Beltran S, Águeda L, Gut M, Gut IG, Distl O. Runs of homozygosity reveal signatures of positive selection for reproduction traits in breed and non-breed horses., 2015, 16: 764.
[32] Druml T, Neuditschko M, Grilz-Seger G, Horna M, Ricard A, Mesaric M, Cotman M, Pausch H, Brem G. Population Networks Associated with Runs of Homozygosity Reveal New Insights into the Breeding History of the Haflinger Horse., 2018, 109(4): 384–392.
[33] Metzger J, Rau J, Naccache F, Bas Conn L, Lindgren G, Distl O. Genome data uncover four synergistic key regulators for extremely small body size in horses., 2018, 19(1): 492.
[34] Beynon SE, Slavov GT, Farré M, Sunduimijid B, Waddams K, Davies B, Haresign W, Kijas J, MacLeod IM, Newbold CJ, Davies L, Larkin DM. Population structure and history of the Welsh sheep breeds determined by whole genome genotyping., 2015, 16: 65.
[35] Muchadeyi FC, Malesa MT, Soma P, Dzomba EF. Runs of homozygosity in Swakara pelt producing sheep: implications on sub-vital performance. In:Proceedings for Association for the Advancement of Animal Breeding and Genetics, 2015, 21: 310–313.
[36] Purfield DC, McParland S, Wall E, Berry DP. The distribution of runs of homozygosity and selection signatures in six commercial meat sheep breeds., 2017, 12(5): e0176780.
[37] Mastrangelo S, Tolone M, Sardina MT, Sottile G, Sutera AM, Di Gerlando R, Portolano B. Genome-wide scan for runs of homozygosity identifies potential candidate genes associated with local adaptation in Valle del Belice sheep., 2017, 49(1): 84.
[38] Guangul SA. Design of community based breeding programs for two indigenous goat breeds of Ethiopia [D]. University of Natural Resources and Life Sciences, 2014.
[39] Onzima RB, Upadhyay MR, Doekes HP, Brito LF, Bosse M, Kanis E, Groenen MAM, Crooijmans RPMA. Genome-Wide characterization of selection signatures and runs of homozygosity in ugandan goat breeds., 2018, 9: 318.
[40] Fleming DS, Koltes JE, Markey AD, Schmidt CJ, Ashwell CM, Rothschild MF, Persia ME, Reecy JM, Lamont SJ. Genomic analysis of Ugandan and Rwandan chicken ecotypes using a 600 k genotyping array., 2016, 17: 407.
[41] Fleming DS, Weigend S, Simianer H, Weigend A, Rothschild M, Schmidt C, Ashwell C, Persia M, Reecy J, Lamont SJ. Genomic comparison of indigenous african and northern european chickens reveals putative mechanisms of stress tolerance related to environmental selection pressure., 2017, 7(5): 1525–1537.
[42] Zhang M, Han W, Tang H, Li G, Zhang M, Xu R, Liu Y, Yang T, Li W, Zou J, Wu K. Genomic diversity dynamics in conserved chicken populations are revealed by genome-wide SNPs., 2018, 19(1): 598.
[43] Ceballos FC, Joshi PK, Clark DW, Ramsay M, Wilson JF. Runs of homozygosity: windows into population history and trait architecture., 2018, 19(4): 220– 234.
[44] Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses., 2007, 81(3): 559–75.
[45] Gusev A, Lowe JK, Stoffel M, Daly MJ, Altshuler D, Breslow JL, Friedman JM, Pe'er I. Whole population, genome-wide mapping of hidden relatedness., 2009, 19(2): 318–26.
[46] Zhang L, Orloff MS, Reber S, Li S, Zhao Y, Eng C. CgaTOH: extended approach for identifying tracts of homozygosity., 2013, 8(3): e57772.
[47] Howrigan DP, Simonson MA, Keller MC. Detecting autozygosity through runs of homozygosity: a comparison of three autozygositydetection algorithms., 2011, 12: 460.
[48] Browning BL, Browning SR. Detecting identity by descent and estimating genotype error rates in sequence data., 2013, 93(5): 840–51.
[49] Magi A, Tattini L, Palombo F, Benelli M, Gialluisi A, Giusti B, Abbate R, Seri M, Gensini GF, Romeo G, Pippucci T. H3M2: detection of runs of homozygosity from whole-exome sequencing data., 2014, 30(20): 2852–2859.
[50] Vigeland MD, Gjøtterud KS, Selmer KK. FILTUS: a desktop GUI for fast and efficient detection of disease- causing variants, including a novel autozygosity detector., 2016, 32(10): 1592–1594.
[51] Narasimhan V, Danecek P, Scally A, Xue Y, Tyler-Smith C, Durbin R. BCFtools/RoH: a hidden Markov model approach for detecting autozygosity from next-generation sequencing data., 2016, 32(11): 1749– 1751.
[52] Szpiech ZA, Blant A, Pemberton TJ. GARLIC: Genomic Autozygosity Regions Likelihood-based Inference and Classification., 2017, 33(13): 2059–2062.
[53] Bjelland DW, Weigel KA, Vukasinovic N, Nkrumah JD. Evaluation of inbreeding depression in Holstein cattle using whole-genome SNP markers and alternative measures of genomic inbreeding., 2013, 96(7): 4697–4706.
[54] Biscarini F, Biffani S, Nicolazzi EL, Morandi N, Stella A. Applying runs of homozygosity to the detection of associations between genotype and phenotype in farm animals. In:Proceedings of the 10th World Congress of Genetics Applied to Livestock Production. Vancouver, BC, Canada, 2014.
[55] Scraggs E, Zanella R, Wojtowicz A, Taylor JF, Gaskins CT, Reeves JJ, de Avila JM, Neibergs HL. Estimation of inbreeding and effective population size of full-blood Wagyu cattle registered with the American Wagyu Cattle Association., 2014, 131(1): 3–10.
[56] Mészáros G, Boison SA, Pérez O'Brien AM, Ferenčaković M, Curik I, Da Silva MV, Utsunomiya YT, Garcia JF, Sölkner J. Genomic analysis for managing small and endangered populations: a case study in Tyrol Grey cattle., 2015, 6: 173.
[57] Williams JL, Hall SJ, Del Corvo M, Ballingall KT, Colli L, Ajmone Marsan P, Biscarini F. Inbreeding and purging at the genomic Level: the Chillingham cattle reveal extensive, non-random SNP heterozygosity., 2016, 47(1): 19–27.
[58] Zavarez LB, Utsunomiya YT, Carmo AS, Neves HH, Carvalheiro R, Ferenčaković M, Pérez O'Brien AM, Curik I, Cole JB, Van Tassell CP, da Silva MV, Sonstegard TS, Sölkner J, Garcia JF. Assessment of autozygosity in Nellore cows (Bos indicus) through high-density SNP genotypes., 2015, 6: 5.
[59] Kim ES, Sonstegard TS, Rothschild MF. Recent artificial selection in U.S. Jersey cattle impacts autozygosity levels of specific genomic regions., 2015, 16: 302.
[60] Iacolina L, Stronen AV, Pertoldi C, Tokarska M, Nørgaard LS, Muñoz J, Kjærsgaard A, Ruiz-Gonzalez A, Kamiński S, Purfield DC. Novel graphical analyses of runs of homozygosity among species and livestock breeds., 2016, 2152847.
[61] Peripolli E, Stafuzza NB, Munari DP, Lima ALF, Irgang R, Machado MA, Panetto JCDC, Ventura RV, Baldi F, da Silva MVGB. Assessment of runs of homozygosity islands and estimates of genomic inbreeding in Gyr (Bos indicus) dairy cattle., 2018, 19: 34.
[62] Forutan M, Ansari Mahyari S, Baes C, Melzer N, Schenkel FS, Sargolzaei M. Inbreeding and runs of homozygosity before and after genomic selection in North American Holstein cattle., 2018, 19(1): 98.
[63] Kim K, Jung J, Caetano-Anollés K, Sung S, Yoo D, Choi BH, Kim HC, Jeong JY, Cho YM, Park EW, Choi TJ, Park B, Lim D, Kim H. Artificial selection increased body weight but induced increase of runs of homozygosity in Hanwoo cattle., 2018, 13(3): e0193701.
[64] Mukherjee A, Mukherjee S, Dhakal R, Mech M, Longkumer I, Haque N, Vupru K, Khate K, Jamir IY, Pongen P, Rajkhowa C, Mitra A, Guldbrandtsen B, Sahana G. High- density genotyping reveals genomic characterization, population structure and genetic diversity of indian Mithun (Bos frontalis)., 2018, 8(1): 10316.
[65] Goszczynski D, Molina A, Terán E, Morales-Durand H, Ross P, Cheng H, Giovambattista G, Demyda-Peyrás S. Runs of homozygosity in a selected cattle population with extremely inbred bulls: descriptive and functional analyses revealed highly variable patterns., 2018, 13(7): e0200069.
[66] Nandolo W, Utsunomiya YT, Mészáros G, Wurzinger M, Khayadzadeh N, Torrecilha RBP, Mulindwa HA, Gondwe TN, Waldmann P, Ferenčaković M, Garcia JF, Rosen BD, Bickhart D, van Tassell CP, Curik I, Sölkner J. Misidentification of runs of homozygosity islands in cattle caused by interference with copy number variation or large intermarker distances., 2018, 50: 43.
[67] Ai H, Huang L, Ren J. Genetic diversity, linkage disequilibrium and selection signatures in chinese and Western pigs revealed by genome-wide SNP markers., 2013, 8(2): e56001.
[68] Silió L, Rodríguez MC, Fernández A, Barragán C, Benítez R, Óvilo C, Fernández AI. Measuring inbreeding and inbreeding depression on pig growth from pedigree or SNP-derived metrics., 2013, 130(5): 349–360.
[69] Zanella R, Peixoto JO, Cardoso FF, Cardoso LL, Biegelmeyer P, Cantão ME, Otaviano A, Freitas MS, Caetano AR, Ledur MC. Genetic diversity analysis of two commercial breeds of pigs using genomic and pedigree data., 2016, 48: 24.
[70] Grossi DA, Jafarikia M, Brito LF, Buzanskas ME, Sargolzaei M, Schenkel FS. Genetic diversity, extent of linkage disequilibrium and persistence of gametic phase in Canadian pigs., 2017, 18(1): 6.
[71] Yang B, Cui L, Perez-Enciso M, Traspov A, Crooijmans RPMA, Zinovieva N, Schook LB, Archibald A, Gatphayak K, Knorr C, Triantafyllidis A, Alexandri P, Semiadi G, Hanotte O, Dias D, Dovč P, Uimari P, Iacolina L, Scandura M, Groenen MAM, Huang L, Megens HJ. Genome-wide SNP data unveils the globalization of domesticated pigs., 2017, 49: 71.
[72] Lago LV, Nery da Silva A, Zanella EL, Groke Marques M, Peixoto JO, da Silva MVGB, Ledur MC, Zanella R. Identification of genetic regions associated with scrotal hernias in a commercial swine herd., 2018, 5(1).
[73] Al-Mamun HA, Clark SA, Kwan P, Gondro C. Genome- wide linkage disequilibrium and genetic diversity in five populations of Australian domestic sheep., 2015, 47: 90.
[74] Kominakis A, Hager-Theodorides AL, Saridaki A, Antonakos G, Tsiamis G. Genome-wide population structure and evolutionary history of the Frizarta dairy sheep., 2017, 11(10): 1680–1688.
[75] Mastrangelo S, Portolano B, Di Gerlando R, Ciampolini R, Tolone M, Sardina MT, International Sheep Genomics Consortium. Genome-wide analysis in endangered populations: a case study in Barbaresca sheep., 2017, 11(7): 1107–1116.
[76] Zhang M, Peng WF, Hu XJ, Zhao YX, Lv FH, Yang J. Global genomic diversity and conservation priorities for domestic animals are associated with the economies of their regions of origin., 2018, 8(1): 11677.
[77] Brito LF, Kijas JW, Ventura RV, Sargolzaei M, Porto-Neto LR, Cánovas A, Feng Z, Jafarikia M, Schenkel FS. Genetic diversity and signatures of selection in various goat breeds revealed by genome-wide SNP markers., 2017, 18(1): 229.
[78] Grossen C, Biebach I, Angelone-Alasaad S, Keller LF, Croll D. Population genomics analyses of European ibex species show lower diversity and higher inbreeding in reintroduced populations., 2018, 11(2): 123– 139.
[79] Zavarez LB, Utsunomiya YT, Carmo AS, Neves HH, Carvalheiro R, Ferenčaković M, Pérez O'Brien AM, Curik I, Cole JB, Van Tassell CP, da Silva MV, Sonstegard TS, Sölkner J, Garcia JF. Assessment of autozygosity in Nellore cows (Bos indicus) through high-density SNP genotypes., 2015, 6: 5.
[80] Visscher PM, Medland SE, Ferreira MA, Morley KI, Zhu G, Cornes BK, Montgomery GW, Martin NG. Assumption- free estimation of heritability from genome-wide identity- by-descent sharing between full siblings., 2006, 2(3): e41.
[81] Peripolli E, Munari DP, Silva MVGB, Lima ALF, Irgang R, Baldi F. Runs of homozygosity: current knowledge and applications in livestock., 2017, 48(3): 255– 271.
[82] Mastrangelo S, Tolone M, Di Gerlando R, Fontanesi L, Sardina MT, Portolano B. Genomic inbreeding estimation in small populations: evaluation of runs of homozygosity in three local dairy cattle breeds., 2016, 10(5): 746–754.
[83] Yang ZC, Huang HT, Yan QX, Wang YC, Yu Y, Chen SH, Sun DX, Zhang SL, Zhang Y. Estimation of genomic inbreeding coefficients based on high-density snp markers in chinese holstein cattle., 2017, 39(1): 416–23.杨湛澄, 黄河天, 闫青霞, 王雅春, 俞英, 陈绍祜, 孙东晓, 张胜利, 张毅. 利用高密度SNP标记分析中国荷斯坦牛基因组近交. 遗传, 2017, 39(1): 16–23.
[84] Msalya G, Kim ES, Laisser EL, Kipanyula MJ, Karimuribo ED, Kusiluka LJ, Chenyambuga SW, Rothschild MF. Determination of genetic structure and signatures of selection in three strains of tanzania shorthorn zebu, boran and friesian cattle by genome-wide snp analyses., 2017, 12(1): e0171088.
[85] VanRaden PM. Efficient methods to compute genomic predictions. J Dairy Sci, 2008, 91(11): 4414–4423.
[86] Garrod AE. Oxon MD, Lond FRCP.The incidence of alkaptonuria: a study of chemical individuality., 1996, 2(3): 274–282.
[87] Szpiech ZA, Xu J, Pemberton TJ, Peng W, Zöllner S, Rosenberg NA, Li JZ. Long runs of homozygosity are enriched for deleterious variation., 2013, 93(1): 90–102.
[88] Huson HJ, Kim ES, Godfrey RW, Olson TA, McClure MC, Chase CC, Rizzi R, O'Brien AM, Van Tassell CP, Garcia JF, Sonstegard TS. Genome-wide association study and ancestral origins of the slick-hair coat in tropically adapted cattle., 2014, 5: 101.
[89] Pryce JE, Haile-Mariam M, Goddard ME, Hayes BJ. Identification of genomic regions associated with inbreeding depression in Holstein and Jersey dairy cattle., 2014, 46: 71.
[90] Bosse M, Megens HJ, Madsen O, Crooijmans RP, Ryder OA, Austerlitz F, Groenen MA, de Cara MA. Using genome-wide measures of coancestry to maintain diversity and fitness in endangered and domestic pig populations., 2015, 25(7): 970–981.
[91] de Cara MÁ, Villanueva B, Toro MÁ, Fernández J. Using genomic tools to maintain diversity and fitness in conservation programmes., 2013, 22(24): 6091– 6099.
[92] Joller S, Bertschinger F, Kump E, Spiri A, von Rotz A, Schweizer-Gorgas D, Drögemüller C, Flury C. Crossed beaks in a local Swiss chicken breed.2018, 14(1): 68.
[93] Li MZ, Zhao YF, Ren J, Jiang SW, Li H. Opportunities and challenges of genetic and breeding research on the livestock in the age of '-omics'., 2017, 39(11): 955–957.李明洲, 赵要风, 任军, 蒋思文, 李辉. 组学时代农业动物遗传育种研究的机遇与挑战. 遗传, 2017, 39(11): 955–957.
[94] Liang SY, Zhou ZK, Hou SS. The research progress of farm animal genomics based on sequencing technologies., 2017, 39(4): 276–292.梁素芸, 周正奎, 侯水生. 基于测序技术的畜禽基因组学研究进展. 遗传, 2017, 39(4): 276–292.
Runs of homozygosity and its application on livestock genome study
Gang Liu, Feizhou Sun, Fangxian Zhu, Haiyong Feng, Xu Han
With the rapid development of high-throughput SNP array and significant reduction of sequencing cost, the techniques of genome-resequencing and SNP chip arrays are widely applied in livestock genomic studies. Long runs of homozygosity (ROH) arose when identical haplotypes were inherited from each parent and thus a long tract of genotypes is homozygous. Nowadays, cumulative studies reported that ROH has progressively served as one of the important indexes to estimate the degree of inbreeding and genetic structure of livestock populations. However, the evaluating criteria of ROH in livestock is still inadequate. In this review, we introduce the history, theory and identification methods of ROH analysis. Meanwhile, we also systematically overview the applications and perspectives of ROH in population genetic structure analysis, genome functional assay, quality investigation and dynamic monitoring of livestock genetic resources.
high-throughput sequencing; runs of homozygosity; population structure; genomic function; genetic defect
2018-10-13;
2019-02-02
畜禽种质资源保护项目(编号:[2018]45)和家养动物平台种质资源项目(编号:2018)资助[Supported by the Protection Project of Animal Germplasm Resources (No. [2018]45) and the National Infrastructure of Domestic Animal Resources (No. 2018)]
刘刚,博士,畜牧师,研究方向:畜禽遗传资源保护与应用。E-mail: lgang-2004@126.com
孙飞舟,博士,研究员,研究方向:畜禽遗传资源保护与应用。E-mail: fzhsun1968@qq.com朱芳贤,高级畜牧师,研究方向:畜禽遗传资源保护与应用。E-mail: 1171277193@qq.com
10.16288/j.yczz.18-287
2019/3/29 9:06:29
URI: http://kns.cnki.net/kcms/detail/11.1913.R.20190329.0906.001.html
(责任编委: 任军)
猜你喜欢
区域治理(2022年40期)2022-11-27
今日农业(2021年11期)2021-08-13
华北电力大学学报(社会科学版)(2021年2期)2021-07-21
中国生殖健康(2020年4期)2020-12-09
中西医结合肝病杂志(2020年2期)2020-10-27
动漫界·幼教365(小班)(2019年10期)2019-10-28
动漫界·幼教365(大班)(2019年10期)2019-10-28
动漫界·幼教365(中班)(2019年10期)2019-10-28
中成药(2018年7期)2018-08-04
图书馆理论与实践(2018年2期)2018-01-28
