
基因组选择在绵羊育种中的应用
2019-04-22 11:54赵志达张莉
遗传 2019年4期
赵志达,张莉
基因组选择在绵羊育种中的应用
赵志达,张莉
中国农业科学院北京畜牧兽医研究所,北京 100193
基因组选择是一种利用高密度芯片全部位点与目的基因存在的连锁不平衡估计基因组育种值的方法,目前已相继在英国、法国、澳大利亚和新西兰等国家的畜禽育种中得到应用并有效提升了育种效率。在我国,基因组选择已在奶牛、生猪和肉鸡的育种中开始应用并取得了一定的成效。我国是世界养羊大国,但在羊的养殖管理、育种水平以及生产效率等方面依旧与发达国家存在较大差距。目前,已有育种工作者尝试对绵羊开展基因组选择育种研究,但至今尚未有比较系统的应用案例。基于绵羊育种基础薄弱的现状,开展基因组选择对我国肉羊产业发展具有重要作用。本文综述了畜禽基因组选择的研究进展及其在绵羊育种中的应用,并对该技术在今后绵羊生产中的指导作用进行了展望。
基因组育种技术;全基因组选择;绵羊育种
畜禽育种通常通过估算育种值(estimated breeding values, EBVs)来预测后代性状,以期得到符合人类需求的产品。EBV的准确度决定了育种工作效率。随着高通量测序技术和生物信息学的发展,利用各种基因组学手段挖掘重要经济性状功能基因已成为畜禽育种的研究热点。目前,已鉴定出一批和畜禽经济性状具有显著相关的数量性状基因座(quantitative trait locus, QTL)。标记辅助选择(marker assisted selection, MAS)就是利用这些已知的QTL进行选育,在一定程度上提高了育种工作效率,但目前所挖掘到的QTL尚不足总量的20%,且MAS难以解释复杂的数量性状,存在一定的局限性。2001年,Meuwissen等[1]首次提出基因组选择(genome selection, GS),该方法使用覆盖全基因组的全部遗传标记进行育种值估计,相比传统方法具有更高的准确性,不仅可以实现早期选种、缩短世代间隔,还有降低近交、加速遗传进展的优点。
目前,GS已成为动植物育种领域的研究热点。我国已在奶牛、生猪和肉鸡中开展,在其他畜禽上应用较少。中国作为养羊大国,对羊及其产品的需求逐年增长,如何选育和培育产量更高、品质更好的优良品种,是当前羊产业发展亟需解决的问题。目前,英国、法国、澳大利亚和新西兰等国家已将GS技术应用于羊的育种中并取得了良好的育种进展,而我国在这方面仍是空白。本文对绵羊()基因组育种技术和GS的发展及应用进行了综述,以期为今后在绵羊上开展GS研究提供借鉴与参考。
1 分子育种技术的发展现状
传统育种主要利用表型信息和系谱信息,通过最佳线性无偏估计(best linear unbiased predication, BLUP)计算育种值,并以此作为畜禽选育的基础从而获得理想的遗传进展。BLUP法在畜禽一些重要经济性状的选择上取得了较好的效果,极大地推动了我国畜禽育种工作。但是通过系谱得出的只是期望的遗传关系,实际中可能因孟德尔抽样离差而失准;同时,对于一些低遗传力、受性别限制、不能早期测定、测定成本高,以及测量难度大的性状,传统育种方法存在较大的局限性。
随着分子遗传学和数量遗传学的发展,畜禽育种逐渐聚焦到分子水平和基因组水平,即基因组育种技术。限制性片段长度多态性技术(restriction fragment length polymorphism, RFLP)于1974年面世,该技术推动了人类对DNA多态性的研究。MAS通过RFLP标记基因组并利用标记的基因型估计性状的表型,以此进行选种。该方法挖掘了与畜禽重要经济性状具有显著相关的QTL或主效基因并进行选种选育,当起始基因位点之间连锁不平衡(linkage disequilibrium, LD)很强时效果更佳,在清除隐性有害基因时尤为明显,并且遗传标记遗传稳定,不易受环境影响,无性别、年龄限制,通过早期选择能够节省成更多的成本[2]。
目前,遗传标记的挖掘手段现已有十几种成熟的技术,如扩增片段长度多态性标记辅助选择(amplified fragment length polymorphism, AFLP)、RFLP、随机扩增多态性标记辅助选择(random amplified polymorphic DNA, RAPD)、微卫星标记辅助选择(simple sequence repeat, SSR)和单核苷酸多态性标记辅助选择(single nucleotide polymorphism, SNP)等。由于SNP是基于单核苷酸水平的多态性,比其他标记遗传稳定性更高,而且因其密度高、分布广、检出率高,更适合快速高通量检测。因此,SNP已成为动植物育种和生命科学研究领域重要工具之一[3]。MAS通过挖掘与重要经济性状显著相关的分子标记,并利用这些标记实现性状的定向选择。该方法需要找到与性状LD的分子标记,但LD状态可能因重组而打破,在不同群体中LD程度亦不相同,所以该方法有时并不可靠。
随着高通量测序技术的发展、商用SNP芯片的问世及分型技术的诞生,测序成本大大降低。全基因组关联分析(genome-wide association study, GWAS)、拷贝数变异(copy number variation, CNV)等方法相继出现,并且用于畜禽经济性状主效基因的挖掘与鉴定。1996年,Risch等[4]提出GWAS方法,该方法以LD为基础,在全基因组水平上识别高密度的SNPs标记并与复杂性状的表型变异进行关联分析,从而分析这些SNPs对表型的遗传效应[5]。GWAS最早应用于人类疾病研究。2005年,杂志首次报道了与年龄具有相关性的黄斑变性GWAS研究[6];之后,又有学者对冠心病[7]、肥胖[8,9]等疾病进行GWAS研究。目前GWAS已广泛应用于挖掘畜禽重要经济性状的遗传标记研究中。张莉等[10]利用50K芯片对329只绵羊(苏尼特羊、德国肉用美利奴羊和杜泊羊)开展体重性状的GWAS分析,检测到与断奶后日增重相关并达到基因组显著水平的SNPs位点10个,达到染色体显著水平的关联位点22个,获得了和等影响绵羊体重性状的重要候选基因。Demars等[11]对Grvette和Olkuska两个绵羊品种进行GWAS分析,得到与绵羊产羔数相关的重要候选基因。Johnston[12]等对Soay绵羊进行GWAS分析,得到控制有无角性状的基因。Doreen等[13]对23只特克赛尔羊的疾病性状进行GWAS分析,发现了影响小眼畸形病的主要候选基因,该基因位于22号染色体上。此外,还有许多针对绵羊多角、毛和毛色、生产发育、繁殖、乳用性状、疾病等不同经济性状的GWAS研究。
CNV是指DNA片段在kb~Mb范围内的突变,包括由插入、缺失、重复和复合多位点导致的变异。2004年,Sebat等[14]提出CNVs多态性在分析与判定人群间的遗传变异上有重要作用。2006年,Redon等[15]发表了人类基因组第一代CNV图谱。由于CNV与某些人类疾病的致病机理存在密切相关性,因此一直是研究的热点。CNV对培育性能优秀、抗病力强的家禽也具有重要意义,刘佳森等[16]利用50K羊芯片对71只苏尼特羊进行基因分型,共检测到134个拷贝数变异区域(copy number variation regions, CNVR),对CNVR覆盖的基因进行注释和功能分析,结果表明这些基因与嗅觉感官知觉、化学刺激感官知觉和识别等环境应答有关,对5个CNVR进行qRT-PCR验证,其中3个CNVR得到验证。侯成林等[17]采用比较基因组杂交芯片技术对3种绵羊(蒙古羊、哈萨克羊和藏羊)进行CNV研究,共检测到28个CNVR,通过对CNVR覆盖的基因进行注释和代谢通路分析,发现在蒙古羊和藏羊的血红蛋白基因存在CNV扩张,该现象可能与两种绵羊高原低氧的生活环境有关。经过验证,83.3%的qRT-PCR结果与芯片检测结果一致。
尽管GWAS和CNV等方法是挖掘畜禽经济性状主效基因的重要方法,但由于GWAS实验结果中存在大量假阳性问题,并且目前研究所得到的具有显著相关的QTL数量有限,同时畜禽大多数重要的经济性状都是受多基因与环境共同调控的复杂的数量性状,因此仅仅依靠以上方法进行选择还存在一定的不足。
2 基因组选择技术及其应用
随着畜禽基因组测序工作的相继完成,测序成本越来越低,加之计算机运算能力不断提升,这为全新育种技术的发展创造了技术条件。GS直接利用全基因组所有的标记效应去估计育种值,为现代育种工作提供了新的思路。
2.1 基因组选择的原理
畜禽性状受一个或多个基因控制,尽管已证实存在一些主效基因,但很多性状的功能基因尚不明确,仍在探索之中。DNA上存在大量的已知位置的SNP标记,它们和某些畜禽重要性状的基因存在一个或多个标记LD。由于每个SNP与主效基因间存在一个或多个LD,多个SNP就可能在不同程度上解释同一个基因的效应,即通过已知SNPs的多态性和表型来估计其效应。假定影响数量性状的每一个QTL和基因芯片中的SNPs存在多个LD[18],通过对全基因组范围内所有与QTL有关的SNP位点进行连锁检测,由参考群的表型估计出每个SNPs的效应,并根据其估计效应对候选群育种值进行估计,由此得到的育种值称为基因组育种值(genomic estimated breeding value, GEBV)[1]。该方法可以追溯到所有影响目标性状的QTL,进一步克服了传统MAS中标记揭示遗传方差较少的问题,提高了育种值的准确性[19],对育种工作起到至关重要的作用。利用SNP分型和SNP效应预测育种值公式:
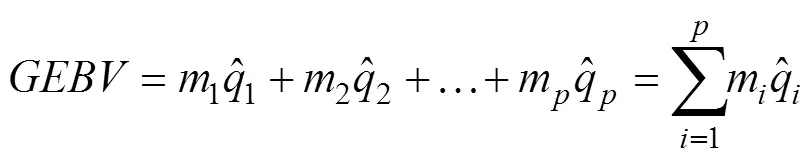
Meuwissen等[1]通过模拟实验得出,仅通过标记预测育种值的准确性就可以达到0.85。2006年,牛全基因组序列信息公布,Schaeffer等[20]对GS在奶牛育种中的应用进行经济学分析,得出GS技术降低了92%的奶牛育种成本。此外,科研人员对GS在猪[21,22]、羊[22,23]和鸡[24~26]等畜禽中的应用策略也进行了探讨。
2.2 基因组育种值的常见算法
2.2.1 间接法
间接法是通过构建参考群,利用参考群个体的表型和全基因组的分型信息,估计全基因组中每一个SNP在不同性状的效应值。之后通过对候选群进行SNPs分型,利用参考群SNPs的标记效应,累加获得候选群的GEBVs (图1)。
间接法常用的估计方法包括岭回归最佳线性无偏预测法(ridge regression best linear unbiased prediction, RRBLUP)和贝叶斯法[1,27,28](BayesA、BayesB、BayesC、BayesS和fBayesB)。
RRBLUP法假设所有标记都有效应,且方差相同;BayesA假设所有标记都有效应,且效应方差服从逆卡方分布(inversed chisquare distribution);BayesB假定标记效应方差以π (π为引入的一个新参数)的概率值为0,以(1-π)的概率服从逆卡方分布[1]。BayesS和BayesA的标记效应方差的分布假设相同,但在效应估计是运用压缩算法[27]。因BayesB计算时间长,Meuwissen等[29]通过条件期望迭代(iteration condition expectation)的算法改进得到fBayesB (fast BayesB)。Verbyla等[30]通过随机搜索变量选择(stochastic search variable selection, SSVS)对π在估计标记效应时进行求解。另外,Meuwissen等[31]将标记效应假定为两个方差不等的正态分布的混合分布,将方差参数和π作为模型变量求解,对BayesB改进后的算法称为BayesC,相较于前者,BayesC计算效率更高。

图1 GS间接法模式计算图
此外,还有主成分分析(principle component analysis)[32]、机器学习(machine learning)[33]、最小二乘回归(least square regression)[1]、半参数法(semiparametric)[34]、非参数法(nonparametric)[35]和贝叶斯LASSO法(least absolute shrinkage and selection operator)[36]等。
2.2.2 直接法
直接法是通过SNPs构建个体间关系矩阵,将关系矩阵放入混合模型方程组(mid model equations, MME)直接获得个体的基因组估计育种值(图2)。
直接法常用的估计方法包括:VanRaden等[37]和Habier等[38]提出的利用标记构建个体间关系矩阵G的基因组最佳线性无偏估计法(genome best linear unbiased prediction, GBLUP);Misztal等[39,40]、Legarra等[41]和Christensen等[42]先后提出的将系谱矩阵A和基因组关系矩阵G构建为H矩阵的一步法(single step GBLUP, SSBLUP);Zhang等[43]提出的给予不同标记不同权重构建关系矩阵的TABLUP法。
2.3 GEBV准确性的影响因素
GEBV的准确性是GEBV和真实育种值(true breeding values, TBV)的相关系数[1,44],计算公式为:
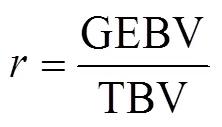
GEBV的准确性直接影响育种工作效率。几种影响GEBV准确性的常见因素如下。
2.3.1 计算模型
对于间接法来说,BayesB方法在多数情况下优于其他方法[1,44],Calus[45]认为这可能与模拟数据中有限的QTL数量有关,与BayesB方法的理论假设比较吻合。Meuwissen等[29]通过对BayesB进行改进得到了计算效率更好的BayesC,fBayesB的准确性比BayesB略低,但运算时间极大缩短。
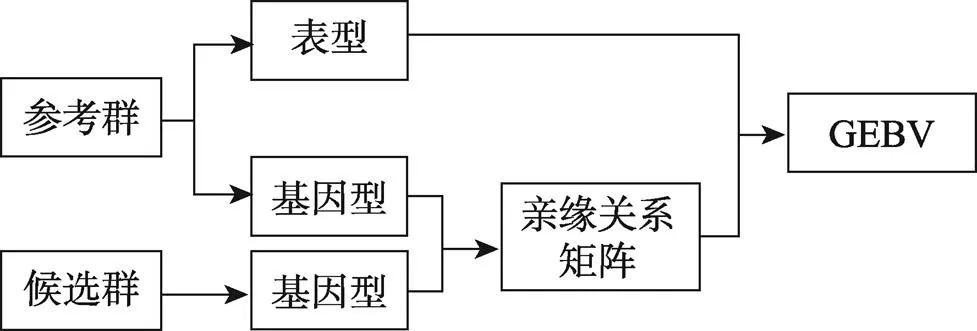
图2 GS直接法模式计算图
对于直接法来说,Goddard等[46]和Hayes等[47]通过理论分析,认为GBLUP和RRBLUP使用相同的信息且假设相同,并得出以上两个方法等价的结论。Zhang等[43]认为,考虑到影响育种值评估时性状的所有基因的位置在全基因组中分布不均匀、基因效应不等的情况,应对标记先进行加权再构建个体关系矩阵,提出了构建TA矩阵(trait-specific relationship matrix, TA矩阵)的TABLUP法。该方法充分考虑了目标性状的遗传结构,因此优于RRBLUP和GBLUP法[48]。
另外,Garrick等[49]认为,对于由少数QTL控制的性状,或来自多品种、远缘相关基因型的数据,Bayes Lasso和MIXTURE算法有效地利用了LD信息,对GEBV的准确性优于GBLUP算法。Ostersen等[50]认为用deregressed EBV代替EBV作为响应变量(response variable),可提高GEBV的准确性,而算法应按照实际情况合理选择。
2.3.2 LD和标记数目
考虑到GS的假设前提为影响数量性状的每一个QTL和基因芯片中的SNPs存在多个LD,即SNPs与QTL必须要存在足够LD,即标记数目越多,准确度越高。Meuwissen等[1]通过类比计算得到相邻标记之间2≥0.2才能进行GS;Calus等[45]则认为2越大,准确度越大,即增加芯片的密度和容量,可提升准确性。但芯片密度越大,会增加成本和计算时长,因此在GS实际工作中,需要充分根据所选性状遗传力的高低和实验成本,选择合适的芯片。Erbe等[51]研究发现,考虑SNP间的LD程度可提升3.6%的准确性,该结果与增加芯片密度结果类似(芯片所含SNP个数由39 745增加到624 214)。此外,利用基因填充技术将低密度填充为中高密度芯片[52,53]和定制个性化芯片,在成本允许的情况下也可以提升GEBV的准确性。
2.3.3 参考群的规模和结构
在对标记效应进行估计时,需要对观察到的表型和基因型进行比较,实际情况中可能会出现各种误差。在概率论中,实验次数越多,频率越趋于概率。因此在减少测定误差的同时,要尽可能记录更多个体的表型信息,即参考群数量越大,标记效应估计越准确。Meuwissen等[1]通过模拟实验得出结论,在对遗传力为0.3的性状进行育种值估计时,样本数大于2000时准确性较高。Goddard等[54]研究表明,性状遗传力越低,所需样本越多,遗传力为0.1的性状需要10 000以上的群体数量才能保证准确度达到0.4。Jeremy等[55]认为,在分析混合性别模型时,应考虑性染色体上的LD,按性别估计GEBV。因此,为了提高GEBV的准确性,需要考虑群体结构,设计足够大的参考群体或选择遗传力较高的目标性状。
2.3.4 非加性效应
大部分重要性状都由多个基因共同调控,基因间的交互作用会影响表型,这种现象称为非加性效应(non-additive allelic effect),主要包括显性效应、上位效应和互作效应,不能稳定遗传。对于非加性效应的计算,Xu等[27,56]通过引入参数,利用近似方法简化计算并在大麦()回交实验中得到应用。Gianola等[34]提出半参数方法并使用模拟数据对一些互作效应进行了研究,认为该方法可能适用于高密度标记数据估计GEBV。
2.3.5 亲缘距离
LD在生殖细胞减数分裂过程中,配子中的DNA可能发生重组或者片段丢失等突变,使SNPs失去与原QTL的LD状态。Meuwissen等[1]通过研究发现,随世代增加,GEBV的准确性会逐渐降低,在经历5个世代后尤为明显,因此每经3个世代就需要重新估计标记效应。Habier等[38]发现随参考群与验证群间世代增加,GEBV的准确性却有所下降。另外,在不同群体间,QTL和SNPs间的LD不尽相同,因此在对同一品种间多场联合测定时,必须要考虑不同场区的LD和环境效应。由此可见,在绵羊实际育种工作中,需要不断完善和优化参考群体。每年可以从基因分型过的候选群中选出一部分进行表型测定,在拥有表型和基因分型时,该群体就可以并入参考群。
2.4 GS技术在羊育种中的应用
目前,国内外在奶牛上实施GS技术已取得明显的遗传进展,但在羊上的应用很少。羊是重要的经济家畜,也是当前我国实施乡村振兴战略和开展精准扶贫的适宜畜种。根据中国畜牧业信息网和FAO (联合国粮食及农业组织,http://www.fao.org)的最新统计,2009~2016年我国绵羊的数量从1.28亿头上升到1.62亿头,羊肉消费量也由433.7万吨增加到468.0万吨,预计今后还会逐年稳步增加。随着羊肉、羊毛、羊奶等羊产品的消费增长,开展羊高效育种和群体改良迫在眉睫,GS技术的实施也是必然的趋势。
羊常规育种分为纯种繁育和杂交育种,其中以杂交育种居多。关于混合品种的GS,Dodds等[57]通过GBLUP计算方法对Romney、Coopworth和Perendale 3种肉毛兼用羊进行GEBV估计与验证,认为GS技术可以在混合品种的群体中进行GEBV估计,该研究考虑了品种结构并得出与预测结果相似的结论,并认为考虑品种结构的效应可以增加GEBV估计的准确度;另外,考虑动物的基因分型会减小偏差,提高准确性。Daetwyler等[58]使用50K芯片运用BayesA和GBLUP两种算法,以纯种美利奴羊和与终端杂交的美利奴羊作为参考群,研究了GS对肉质和羊毛预测的准确性,其中GBLUP和BayesA结论差异明显,说明当前SNP密度不足以跨物种使用标记效应,可能由不同品种中相同QTL的位置不同导致;该研究认为随着参考群样本含量以及SNP芯片密度增加,将会进一步提高GEBV的准确性。
繁殖性状直接影响养羊的经济效益,是育种中需要重点关注的经济性状,但由多基因控制且受环境影响,遗传力极低。Newton等[59]利用基因组信息评估澳大利亚绵羊繁殖性状的遗传进展,研究发现不同年龄的基因组信息会影响遗传增益,公羊1岁时的遗传增益显著高于2岁。Pickering等[60]对4237只Romney羊进行GS预测,发现繁殖性状的准确性在0.16~0.52之间。
胴体性状和羊毛性状也是绵羊的重要经济性状。Auvray等[61]对Romney、Coopworth、Perendale绵羊进行GS分析,并得出GS技术可以在以上3种羊中进行断奶重、胴体重和粪便虫卵数的育种值预测,在Romney中可以对产羔数进行预测。Slack- Smith等[62]研究发现,GS在澳大利亚绵羊胴体、生长性状、冻后眼肌面积和肌间脂肪含量的准确性分别为0.71、0.82、0.78和0.61。Brito等[63]在新西兰绵羊活体性状准确性范围为(0.18±0.07)~(0.33±0.10),胴体性状为(0.28±0.09)~(0.55±0.05),肉质性状为(0.21±0.07)~ (0.36±0.08)。Moghaddar等[64]对Poll Dorset、White Suffolk和Border Leicester 3种羊的体重进行GEBV估计,Poll Dorset 的估计准确性为0.11~0.27,后两个品种的准确性在0.25~0.63之间。Shumbusho等[65]认为,在优化育种方案或使用全基因组信息均比传统方法得到更高的绵羊产肉性状遗传度进展。Daetwyler等[66]指出,仅针对纯种美利奴羊来说,1岁时的油性羊毛重量(greasy fleece weigh, GFW)和1岁时羊毛纤维直径(fibre diameter, FD)的准确度均超过0.70,短纤维强度(staple strength, SS)的准确性低于0.70,这可能与SS记录的数量少或者SS比GFW和FD的遗传力更低相关。Moghaddar等[64]发现GS对1岁和成年的Merino羊毛性状的估计准确性为0.33~0.75。Bolormaa等[67]对3种羊毛质量性状进行GS分析,发现BayesR和GBLUP算法的平均GEBV精确度相似,约为0.22,BayesR对羊毛产量和FD准确度均大于0.40,而对羊皮质量和污浊程度的准确度较差。
羊的泌乳性状与羔羊的成活率密切相关,且羊奶也具有重要的经济价值,因此对羊泌乳性状和乳房性状的研究也极为重要。Duchemin等[68]对法系Lacaune绵羊群体(约2500只)的泌乳性状进行了研究,认为GS技术可以使公羔的GEBV精确度提升18%~25%,BayesC计算时间过长,sPLS法对于GEBV的估计具有稳健性,其结果非常接近GBLUP结果。McLaren等[69]通过分析29个群体2957头纯种Texel母羊,认为其乳房形态和乳房炎有较高的相关性,并影响羊肉产量,该研究认为可以利用GS技术通过分析影响乳房形态的基因提高羊肉产量,并认为这是肉羊育种的一种新思路。Mucha等[70]对1960只英国奶山羊的产奶量和泌乳量进行GS试验,得出SSBLUP和GBLUP的预测准确性分别为0.61和0.32,SSBLUP具有更高的准确性。Carillier等[71]对法国Saanen奶山羊和Alpine羊进行GS选择育种,GEBV交叉验证的准确性为36%~53%。Larroque等[72]研究发现,GBLUP在奶山羊和奶绵羊上的准确性比荷斯坦奶牛差,但SSBLUP的准确性比GBLUP高,通过交叉验证发现,法国Lacaune奶绵羊GEBV准确性增加了0.47,Lacaune奶山羊增加了0.43。Baloche等[73]通过对比BLUP和SSBLUP对Lacaune奶山羊的选择效果,发现GS比传统育种方法的准确性更高,达到52%;此外,SSBLUP比BLUP准确性高58%。Molina等[74]使用55K芯片估计西班牙Florida奶山羊的GEBV,结果显示SSBLUP平均增加5.86%的准确性。
在验证不同设计模型中GS的效益研究中,Van der Werf等[23]在肉羊和细毛羊群体中进行GS模拟实验,结果表明,相比传统方法,GS方法在肉羊和细毛羊中经济选择指数分别提高了30%和40%。Raoul等[75]采用低密度SNP芯片(very low-density SNP panel)对Lacaune绵羊进行GS,结果表明:对雄性参考群使用中等密度基因型(medium-density genotypes)时,基因组设计(genomic design)的遗传效益提升26%,对同时包含雄性和雌性的参考群使用中等密度的基因型,其遗传效益提升54%;无论基因组情况如何,基因组设计的近亲繁殖率都会比传统设计低。对雌性和雄性候选绵羊采用极低密度的基因型与归集过程(imputation process)相结合导致小型的绵羊育种计划的遗传效益大幅增加。Santos等[76]通过比较不同的绵羊育种策略的准确性及经济收益,认为显性+选择性基因组选择(pheno + selective GS)可以在最短时间内实现盈亏平衡且效益最大,并有效降低了遗传间隔。
3 结语与展望
GS育种比传统育种技术具有更高的准确性,可实现早期选种、缩短世代间隔,还具有降低近交、加速遗传进展等优点,成为国内外畜禽育种工作的研究热点[77]。在我国,GS已在奶牛、生猪和肉鸡上大量应用,一些企业和实验室开始对绵羊育种进行GS探索,但至今尚未有系统的研究和结果。国外GS的案例已经证明该技术能显著提高绵羊的生长性状、羊毛性状和泌乳性状等选择的准确性。也有研究表明在考虑基因分型、增加样本量和群体构成时可获得更高的准确性,而采用极低密度芯片和归集过程相结合、显性和选择性GS相结合也可以增加遗传效益。
GS技术对畜禽育种的贡献很大,但在理论研究和实际操作中也存在一些问题,主要表现在以下4个方面:(1) GS需要种羊场有完整的系谱、生产性能测定资料,同时需要持续的测定,测定费用高,回报慢;(2) GS对低遗传力性状选择效果较差,需要大规模的参考群;(3) GS依据参考群的SNPs效应估计候选群的GEBV,但不适用与新出现的精英个体和远缘个体[55];(4)计算时间较长,仍需进一步研究算法。
目前,生长速度慢、繁殖效率低是我国肉羊产业发展中的瓶颈问题,因此,在制定育种目标时应结合产业的需求进行合理的选育。生产大数据是开展GS研究的基础,羊场必须要持续开展生产性能测定并建立核心群生产信息数据平台。为降低GS成本,可以先采用传统方法进行选择,然后再进行SSBLUP选择。以某羊场为例,假设对3000只候选羊进行GS选育,育种目标是提高生长速度和增加产羔数,可以先用BLUP对候选群体进行育种值估计并排名,然后对排名前30%(选择强度视实际情况)的个体再进行GEBV估计。对这批羊进行后续生长性能测定,得到表型+系谱+基因型信息,该群体又可并入参考群中(节省参考群测定费用)。此外通过和其他相同品种的羊场进行联合育种[78],羊场之间可以互相导血,使开展联合育种的羊存在一定的亲缘关系,既可使联合场区的参考群的信息得到共享(另需考虑场区效应),同时还扩大了参考群的规模,提高了选择的准确性和效率,并极大程度地降低了测定成本。同时,还应结合对候选群体的表型选择,以减少精英个体的遗漏。此外,我国羊品种资源丰富,自主研发覆盖与绵羊重要经济性状相关联位点的中低密度育种芯片,亦能提高GBEV的准确性。
BLUP和MAS在羊育种工作中虽取得一定的成效,但面临由多基因控制的复杂数量性状时,只能在很小的层面去解释,而GS通过关联基因组内所有标记进行位点效应估计,很好地解决了传统方法难以解决的问题,是今后畜禽育种的大趋势,也是育种发展史上的一个重要里程碑。中国农业科学院北京畜牧兽医研究所对肉鸡和肉牛进行了GS研究并获得较好的成效,同时自主研发出鸡芯片“IASCHICK”。本研究团队将对已有的绵羊育种结果进行梳理和总结,研发更加适合我国绵羊育种的基因芯片,并开展湖羊的GS育种研究,探索GS技术在羊育种中的方法和效率。
[1] Meuwissen THE, Hayes BJ, Goddard ME. Prediction of total genetic value using Genome-Wide dense marker maps., 2001, 157(4): 1819–1829.
[2] Lu SX, Wu CX. Reseach and application of animal genetic marker-assisted selection.,2002, 24(3): 359–362.鲁绍雄, 吴常信. 动物遗传标记辅助选择研究及其应用. 遗传, 2002, 24(3): 359–362.
[3] Tang LQ, Xiao CL, Wang WP. Progress in research and application of SNP molecular markers.2012, 28(12): 154–158.唐立群, 肖层林, 王伟平. SNP分子标记的研究及其应用进展. 中国农学通报, 2012, 28(12): 154–158.
[4] Risch N, Merikangas K. The future of genetic studies of complex human diseases., 1996, 273(5281): 1516–1517.
[5] Hirschhorn JN, Daly MJ. Genome-wide association studies for common diseases and complex traits., 2005, 6(2): 95–108.
[6] Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, Henning AK, SanGiovanni JP, Mane SM, Mayne ST, Bracken MB, Ferris FL, Ott J, Barnstable C, Hoh J. Complement factor H polymorphism in Age-Related macular degeneration., 2005, 308(5720): 385–389.
[7] Samani NJ, Erdmann J, Hall AS, Hengstenberg C, Mangino M, Mayer B, Dixon RJ, Meitinger T, Braund P, Wichmann HE, Barrett JH, König IR, Stevens SE, Szymczak S, Tregouet D, Iles MM, Pahlke F, Pollard H, Lieb W, Cambien F, Fischer M, Ouwehand W, Blankenberg S, Balmforth AJ, Baessler A, Ball SG, Strom TM, Braeænne I, Gieger C, Deloukas P, Tobin MD, Ziegler A, Thompson JR, Schunkert H, WTCCC and the Cardiogenics Consortium. Genomewide association analysis of coronary artery disease., 2007, 357(5): 443–453.
[8] Herbert A, Gerry NP, Mcqueen MB, Heid IM, Pfeufer A, Illig T, Wichmann HE, Meitinger T, Hunter DJ, Hu FBJS. A common genetic variant is associated with adult and childhood obesity., 2006, 312(5771): 279–283.
[9] Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, Perry JRB, Elliott KS, Lango H, Rayner NW, Shields B, Harries LW, Barrett JC, Ellard S, Groves CJ, Knight B, Patch AM, Ness AR, Ebrahim S, Lawlor DA, Ring SM, Ben-Shlomo Y, Jarvelin MR, Sovio U, Bennett AJ, Melzer D, Ferrucci L, Loos R JF, Barroso I, Wareham NJ, Karpe F, Owen KR, Cardon LR, Walker M, Hitman GA, Palmer CN, Doney AS, Morris AD, Smith GD, Hattersley AT, McCarthy MI. A common variant in the fto gene is associated with body mass index and predisposes to childhood and adult obesity., 2007, 316(5826): 889–894.
[10] Zhang L, Liu JS, Xu LY, Zhao FP, Lu J, Zhang SF, Wang HH, Zhang XN, Wei CH, Lu GB, Zheng YM, Du LX. Genome-wide association analysis of sheep weight traits., 2014, (2): 194–203.张莉, 刘佳森, 徐凌洋, 赵福平, 陆健, 张世芳, 王慧华, 张晓宁, 魏彩虹, 陆国彬, 郑友民, 杜立新. 绵羊体重性状全基因组关联分析. 中国畜牧兽医, 2014, (2): 194–203.
[11] Demars J, Fabre S, Sarry J, Rossetti R, Gilbert H, Persani L, Tosser-klopp G, Mulsant P, Nowak Z, Drobik W, Martyniuk E, Bodin L. Genome-Wide association studies identify two novel BMP15 mutations responsible for an atypical hyperprolificacy phenotype in sheep., 2013, 9(4): e1003482.
[12] Johnston SE, Mcewan JC, Pickering NK, Kijas JW, Dario B, Pilkington JG, Pemberton JM, Slate J. Genome-wide association mapping identifies the genetic basis of discrete and quantitative variation in sexual weaponry in a wild sheep population., 2011, 20(12): 2555–2566.
[13] Becker D, Tetens J, Brunner A, Bürstel D, Ganter M, Kijas J, International Sheep Genomics Consortium, Drögemüller C. Microphthalmia in Texel sheep is associated with a missense mutation in the paired-like homeodomain 3 (PITX3) gene., 2010, 5(1): e8689.
[14] Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, Månér S, Massa H, Walker M, Chi M, Navin N, Lucito R, Healy J, Hicks J, Ye K, Reiner A, Gilliam TC, Trask B, Patterson N, Zetterberg A, Wigler M. Large-Scale copy number polymorphism in the human genome., 2004, 305(5683): 525–528.
[15] Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, Cho EK, Dallaire S, Freeman JL, González JR, Gratacòs M, Huang J, Kalaitzopoulos D, Komura D, MacDonald J R, Marshall CR, Mei R, Montgomery L, Nishimura K, Okamura K, Shen F, Somerville MJ, Tchinda J, Valsesia A, Woodwark C, Yang F, Zhang J, Zerjal T, Zhang J, Armengol L, Conrad DF, Estivill X, Tyler-Smith C, Carter NP, Aburatani H, Lee C, Jones KW, Scherer SW, Hurles ME. Global variation in copy number in the human genome., 2006, 444(7118): 444–454.
[16] Liu JS, Zhang L, Li YH, Zhao FP, Wei CH, Du LX. Study on genomic distribution characteristics of copy number variation of Sunite sheep., 2013, (10): 173–178.刘佳森, 张莉, 李蕴华, 赵福平, 魏彩虹, 杜立新. 苏尼特羊拷贝数变异的基因组分布特征研究. 中国畜牧兽医, 2013, (10): 173–178.
[17] Hou CL, Wang W, Zhou HM, Zhang YR, Cao JW. Study on copy number variation of sheep genomes in northern China., 2016, 43(3): 784–790.侯成林, 王伟, 周欢敏, 张焱如, 曹俊伟. 中国北方绵羊基因组拷贝数变异研究. 中国畜牧兽医, 2016, 43(3): 784–790.
[18] Goddard ME, Hayes BJ. Genomic selection., 2015, 124(6): 323–330.
[19] Vanraden PM, Van Tassell CP, Wiggans GR, Sonstegard TS, Schnabel RD, Taylor JF, Schenkel FS. Invited review: reliability of genomic predictions for North American Holstein bulls., 2009, 92(1): 16–24.
[20] Schaeffer LR. Strategy for applying genome-wide selection in dairy cattle., 2006, 123(4): 218– 223.
[21] Tuggle CK, Dekkers JCM. Genotyping: how useful is it for producers?, 2009, 25(9): 21–23.
[22] Simianer H. The potential of genomic selection to improve litter size in pig breeding programs.In: Proceedings 60th Annual Meeting of the European Association of Animal Production. Barcelona, Spain, 2009.
[23] Van der Werf JHJ. Potential benefit of genomic selection in sheep.In: Proceedings of the Association for the Advancement of Animal Breeding and Genetics. Barossa Valley, South Australia, 2009.
[24] Wolc A, Stricker C, Arango J, Settar P, Fulton JE, O'Sullivan NP, Preisinger R, Habier D, Fernando R, Garrick DJ, Lamont S, Dekkers JC. Breeding value prediction for production traits in layer chickens using pedigree or genomic relationships in a reduced animal model., 2011, 43(1): 5.
[25] Chen CY. Genome wide marker assisted selection in chicken: making the most of all data, pedigree, phenotypic, and genomic in a simple one step procedure., 2010, 22(1): 22.
[26] Legarra A, Calenge F, Mariani P, Velge P, Beaumont C. Use of a reduced set of single nucleotide polymorphisms for genetic evaluation of resistance to Salmonella carrier state in laying hens., 2011, 90(4): 731–736.
[27] Xu S. Estimating polygenic effects using markers of the entire genome., 2003, 163(2): 789–801.
[28] Wang CL, Ding XD, Liu JF, Yin ZJ, Zhang Q. Bayesian methods for genomic breeding value estimation., 2014, 36(2): 111–118.王重龙, 丁向东, 刘剑锋, 殷宗俊, 张勤. 基因组育种值估计的贝叶斯方法. 遗传, 2014, 36(2): 111–118.
[29] Meuwissen TH, Solberg TR, Shepherd R, Woolliams JA. A fast algorithm for BayesB type of prediction of genome- wide estimates of genetic value., 2009, 41(1): 2.
[30] Verbyla KL, Hayes BJ, Bowman PJ, Goddard ME. Accuracy of genomic selection using stochastic search variable selection in Australian Holstein Friesian dairy cattle., 2009, 91(5): 307–311.
[31] Meuwissen TH. Accuracy of breeding values of 'unrelated' individuals predicted by dense SNP genotyping., 2009, 41(1): 35.
[32] Solberg TR, Sonesson AK, Woolliams JA, Meuwissen TH. Reducing dimensionality for prediction of genome-wide breeding values., 2009, 41(1): 29.
[33] Long N, Gianola D, Rosa GJ, Weigel KA, Avendano S. Machine learning classification procedure for selecting SNPs in genomic selection: application to early mortality in broilers., 2008, 132: 373–376.
[34] Gianola D, Fernando RL, Stella A. Genomic-Assisted prediction of genetic value with semiparametric procedures., 2006, 173(3): 1761–1776.
[35] Bennewitz J, Solberg T, Meuwissen T. Genomic breeding value estimation using nonparametric additive regression models., 2009, 41(1): 20.
[36] de los Campos G, Naya H, Gianola D, Crossa J, Legarra A, Manfredi E, Weigel K, Cotes JM. Predicting quantitative traits with regression models for dense molecular markers and pedigree., 2009, 182(1): 375–385.
[37] Vanraden PM. Efficient methods to compute genomic predictions., 2008, 91(11): 4414–4423.
[38] Habier D, Tetens J, Seefried FR, Lichtner P, Thaller G. The impact of genetic relationship information on genomic breeding values in German Holstein cattle., 2010, 42(1): 5.
[39] Misztal I, Legarra A, Aguilar I. Computing procedures for genetic evaluation including phenotypic, full pedigree, and genomic information., 2009, 92(9): 4648– 4655.
[40] Aguilar I, Misztal I, Johnson DL, Legarra A, Tsuruta S, Lawlor TJ. Hot topic: a unified approach to utilize phenotypic, full pedigree, and genomic information for genetic evaluation of Holstein final score., 2010, 93(2): 743–752.
[41] Legarra A, Ducrocq V. Computational strategies for national integration of phenotypic, genomic, and pedigree data in a single-step best linear unbiased prediction., 2012, 95(8): 4629–4645.
[42] Christensen OF, Lund MS. Genomic prediction when some animals are not genotyped., 2010, 42(1): 2–2.
[43] Zhang Z, Ding XD, Liu JF, De Koning DJ, Zhang Q. TA-BLUP: A new genetic evaluation method for genomic selection. In: The Proceedings of the 9th World Congress on Genetics Applied to Livestock Production (WCGALP). Leizig, GermatW, 2010.
[44] Zhang Z, Zhang Q, Ding XD. Advances in genomic selection in domestic animals., 2011, 56(25): 2655–2663.
[45] Calus MP. Genomic breeding value prediction: methods and procedures., 2010, 4(2): 157–164.
[46] Goddard M. Genomic selection: prediction of accuracy and maximisation of long term response., 2009, 136(2): 245–257.
[47] Hayes BJ, Visscher PM, Goddard ME. Increased accuracy of artificial selection by using the realized relationship matrix., 2009, 91(1): 47–60.
[48] Zhang Z, Liu J, Ding X, Bijma P, de Koning DJ, Zhang Q. Best linear unbiased prediction of genomic breeding values using a trait-specific marker-derived relationship matrix., 2010, 5(9): e12648.
[49] Garrick DJ, Taylor JF, Fernando RL. Deregressing estimated breeding values and weighting information for genomic regression analyses.,2009, 41(1): 55.
[50] Ostersen T, Christensen OF, Henryon M, Nielsen B, Su G, Madsen P. Deregressed EBV as the response variable yield more reliable genomic predictions than traditional EBV in pure-bred pigs., 2011, 43(1): 38.
[51] Erbe M, Hayes BJ, Matukumalli LK, Goswami S, Bowman PJ, Reich CM, Mason BA, Goddard ME. Improving accuracy of genomic predictions within and between dairy cattle breeds with imputed high-density single nucleotide polymorphism panels., 2012, 95(7): 4114–4129.
[52] Weigel KA, Van Tassell CP, O’Connell JR, Vanraden PM, Wiggans GR. Prediction of unobserved single nucleotide polymorphism genotypes of Jersey cattle using reference panels and population-based imputation algorithms., 2010, 93(5): 2229–2238.
[53] Zhang Z, Druet T. Marker imputation with low-density marker panels in Dutch Holstein cattle., 2010, 93(11): 5487–5494.
[54] Goddard ME, Hayes BJ. Mapping genes for complex traits in domestic animals and their use in breeding programmes., 2009, 10(6): 381–391.
[55] Taylor JF. Implementation and accuracy of genomic selection., 2014, 420–421: S8–S14.
[56] Xu S, Jia Z. Genomewide analysis of epistatic effects for quantitative traits in barley., 2007, 175(4): 1955–1963.
[57] Dodds KG, Auvray B, Lee M, Newman S, McEwan JC. Genomic selection in New Zealand dual purpose sheep. In: 10th World Congress on Genetics Applied to Livestock Production. Vancouver, Canada, 2014.
[58] Daetwyler HD, Kemper KE, van der Werf JHJ, Hayes BJ. Components of the accuracy of genomic prediction in a multi-breed sheep population1., 2012, 90(10): 3375–3384.
[59] Newton JE, Brown DJ, Dominik S, van der Werf JHJ. Impact of young ewe fertility rate on risk and genetic gain in sheep-breeding programs using genomic selection., 2017, 57(8): 1653–1664.
[60] Pickering NK, Dodds KG, Auvray B, Mcewan JC, Villalobos NL. The impact of genomic selection on genetic gain in the New Zealand sheep dual purpose selection index. In: Proceedings of the Association Advancement Animal Breeding and Genetics. Napier, New Zealand, 2013.
[61] Auvray B, McEwan JC, Newman S, Lee M, Dodds KG. Genomic prediction of breeding values in the New Zealand sheep industry using a 50K SNP chip., 2014, 92(10): 4375–4389.
[62] Slack-Smith A, Kinghorn BP, van der Werf JHJ. Accuracy of genomic selection in predicting carcass traits in meat sheep., 2010, 50(11–12): XIII–XIII.
[63] Brito LF, Clarke SM, Mcewan JC, Miller SP, Pickering NK, Bain WE, Dodds KG, Sargolzaei M, Schenkel FS. Prediction of genomic breeding values for growth, carcass and meat quality traits in a multi-breed sheep population using a HD SNP chip., 2017, 18(1): 7.
[64] Moghaddar N, Swan AA, van der Werf J HJ. Genomic prediction of weight and wool traits in a multi-breed sheep population., 2014, 54(5): 544–549.
[65] Shumbusho F, Raoul J, Astruc JM, Palhiere I, Elsen JM. Potential benefits of genomic selection on genetic gain of small ruminant breeding programs., 2013, 91(8): 3644–3657.
[66] Daetwyler HD, Hickey JM, Henshall JM, Dominik S, Gredler B, van der Werf JHJ, Hayes BJ. Accuracy of estimated genomic breeding values for wool and meat traits in a multi-breed sheep population., 2010, 50(12): 1004–1010.
[67] Bolormaa S, Swan AA, Brown DJ, Hatcher S, Moghaddar N, van der Werf JH, Goddard ME, Daetwyler HD. Multiple-trait QTL mapping and genomic prediction for wool traits in sheep., 2017, 49(1): 62.
[68] Duchemin SI, Colombani C, Legarra A, Baloche G, Larroque H, Astruc JM, Barillet F, Robert-Granié C, Manfredi E. Genomic selection in the French Lacaune dairy sheep breed., 2012, 95(5): 2723–2733.
[69] Mclaren A, Kaseja K, Yates J, Mucha S, Lambe NR, Conington J. New mastitis phenotypes suitable for genomic selection in meat sheep and their genetic relationships with udder conformation and lamb live weights., 2018, 12(12): 2470–2479.
[70] Mucha S, Mrode R, Maclaren-Lee I, Coffey M, Conington J. Estimation of genomic breeding values for milk yield in UK dairy goats., 2015, 98(11): 8201–8208.
[71] Carillier C, Larroque H, Palhière I, Clément V, Rupp R, Robert-Granié C. A first step toward genomic selection in the multi-breed French dairy goat population., 2013, 96(11): 7294–7305.
[72] Larroque H, Barillet F, Baloche G, Astruc J M, Buisson D, Shumbusho F, Clment V, Lagriffoul G, Palhire I, Rupp R. Toward genomic breeding programs in French dairy sheep and goats. In: 10th World Congress on Genetics Applied to Livestock Production. Vancouver, Canada, 2014.
[73] Baloche G, Astruc JM, Boulenc P, Giralviala B, Guibert P, Panis P, Legarra A, Barillet F. Genomic selection experiment in Lacaune dairy sheep: Progeny test results of rams initially selected either on parent average or on genomic prediction. In: 10th World Congress on Genetics Applied to Livestock Production. Vancouver, Canada, 2014.
[74] Molina A, Muñoz E, Díaz C, Menéndez-Buxadera A, Ramón M, Sánchez M, Carabaño MJ, Serradilla JM. Goat genomic selection: Impact of the integration of genomic information in the genetic evaluations of the Spanish Florida goats., 2018, 163: 72–75.
[75] Raoul J, Swan AA, Elsen JM. Using a very low-density SNP panel for genomic selection in a breeding program for sheep., 2017, 49(1): 76.
[76] Santos BFS, van der Werf JHJ, Gibson JP, Byrne TJ, Amer PR. Genetic and economic benefits of selection based on performance recording and genotyping in lower tiers of multi-tiered sheep breeding schemes., 2017, 49(1): 10.
[77] Tan C, Bian C, Yang D, Li N, Wu ZF, Hu XX. Application of genomic selection in farm animal breeding., 2017, 39(11): 1033–1045.谈成, 边成, 杨达, 李宁, 吴珍芳, 胡晓湘, 李明洲. 基因组选择技术在农业动物育种中的应用. 遗传, 2017, 39(11): 1033–1045.
[78] Li HD, Bao ZM, Sun XW. Genomic selection and its application., 2011, 33(12): 1308–1316.李恒德, 包振民, 孙效文. 基因组选择及其应用. 遗传, 2011, 33(12): 1308–1316.
Applications of genome selection in sheep breeding
Zhida Zhao, Li Zhang
Genome selection is a method for estimating genomic breeding values using linkage disequilibrium between all sites of high-density chips and target genes. Genome selection has been applied to enhance livestock breeding efficiency in countries such as UK, France, Australia and New Zealand. In China, this technique has been successfully practiced in dairy cattle, swine, and broiler, and achieved significant progress. China raises the majority of sheep throughout the world. However, a large technological gap in sheep breeding and production efficiency exists between China and other developed countries. Some breeders have attempted to use genome selection in sheep breeding; however, there are no good cases till now. Based on the current level of animal breeding, the genome selection method should play an important role in the future of the meat sheep industry. In this review, we summarize the research of genome selection in livestock and poultry as well as some applications in sheep breeding. We also describe the prospects for the future application of this technology to improve the efficiency, quality and outcomes of sheep production.
genomic breeding technology; genomic selection; sheep breeding
2018-11-14;
2019-02-05
中央级公益性科研院所基本科研业务费专项(编号:Y2017XM02)资助[Supported by the Central Public-interest Scientific Institution Basal Research Fund (No. Y2017XM02)]
赵志达,硕士研究生,专业方向:羊分子育种及生产。E-mail: 707187879@qq.com
张莉,博士,研究员,研究方向:畜禽分子遗传育种及生产,畜禽遗传资源保存和评价。E-mail: zhangli07@caas.cn
10.16288/j.yczz.18-251
2019/4/1 14:01:37
URI: http://kns.cnki.net/kcms/detail/11.1913.R.20190401.1401.003.html
(责任编委: 蒋思文)
猜你喜欢
今日农业(2021年11期)2021-08-13
建材发展导向(2021年10期)2021-07-16
新世纪智能(语文备考)(2021年11期)2021-03-08
中国生殖健康(2020年4期)2020-12-09
中西医结合肝病杂志(2020年2期)2020-10-27
意林(儿童绘本)(2019年6期)2019-08-05
小学生必读(低年级版)(2019年12期)2019-04-20
中成药(2018年7期)2018-08-04
幼儿画刊(2018年7期)2018-07-24
中国公路(2017年11期)2017-07-31
