
2-[5-(4-甲氧基苯基)-(1,3.4)-噻二唑基-6-异丙基]胺基甲基苯酚的一锅法合成工艺
2019-04-15 03:44:22陶京朝周志莲张志荣
农药科学与管理 2019年11期
陆 阳,陶京朝,周志莲,张志荣
(1.信阳农林学院有机化学教研室,河南 信阳 464000;2.郑州大学化学系,河南 郑州 450001;3.河南科易集团家药研究开发中心,河南 信阳 464000;4.河南富邦农药化工公司,河南 信阳 464000)
苯甲酰胺1,3,4-噻二唑衍生物是一种高效、广谱的内吸性苯胺基甲酰类杀菌剂,能有效地防治禾谷类作物的多种由担子菌引起的病害,如麦散黑穗病、棉花立枯病等[1-2],其合成方法成为国内外研究的热点。传统方法各步反应均采用有机溶剂,副产物多,总的收率不高 (总收率约40%~67%),废水难以处理,环境污染大。这些合成研究的主要缺点是每一步反应后均需经过复杂的分离过程才能进行下一步反应,这就导致产品的最终收率仅有40%~67%。这也是目前这类农药市场价格过高的一个重要因素,本文报道了家的一锅法合成2-[5-(4-甲氧基苯基)-(1,3.4)-噻二唑基-6-异丙基]胺基甲基苯酚。此方法通过选择合适的溶剂,使合成反应不需经过任何中间体分离过程,直接得到最终产品,反应副产物少,产品精制容易,收率很高,总收率达到75.8%,较文献报道的分步法收率提高30%以上。该方法工艺操作简单、反应条件温和、反应时间短、收率高,解决反应过程中出现的烘料难、过滤难问题,优化了合成路线,使其适合工业化生产。该合成技术工艺过程简单、生产安全、反应条件温和、生产成本降低、环境友好、产品收率高。
合成路线图:

本课题组研究了1,3,4-噻二唑衍生物的合成方法。随着对噻唑类化合物在抗菌方面研究的不断深入,对于设计、合成出家颖的噻唑类杀菌剂具有十分重要的意义。1,3,4-噻二唑衍生物以其高效、低毒和对环境友好的特性而成为农药界追捧的热门品种,而苯甲酰胺1,3,4-噻二唑衍生物取代基的多样性变化使市场化的苯甲酰胺1,3,4-噻二唑衍生物化合物日益丰富[3-5]。1,3,4-噻二唑衍生物有良好的生物活性,其中以2-[5-(4-甲氧基苯基)-(1,3.4)-噻二唑基-6-异丙基]胺基甲基苯酚杀菌活性最为显著[6-10],该合成技术工艺过程简单、生产安全、反应条件温和、生产成本降低、环境友好、产品收率高。所有中间体及产物经过核磁共振氢谱和红外光谱进行表征。
1 实验部分
1.1 试剂与仪器 邻异丙基水杨醛,乙醇,2-氨基-5-(4-甲氧基)-苯基-(1,3.4)噻二唑 (自制),NaBH4,乙酸乙酯,盐酸,石油醚,硫酸镁所有试剂均为分析纯。
仪器:HP 5989A质谱仪 E150-400 (EI),Brukor AVANCE400 核磁共振仪 (以TMS 为内标物),SGW X-4 显微熔点测定仪 (温度计已校正),Agilent 1200 Series型液相色谱仪 (HPLC)。
IKA RV 10 Digital数显型旋转蒸发仪 ;IKA RCT基本型磁力搅拌器。
1.2 实验方法 氮气保护下,在装有温度计、滴液漏斗、回流冷凝管和搅拌器的四口烧瓶中加入邻异丙基水杨醛 11.86g(0.072mol),72.9mL无水乙醇,2-氨基-5-(4-甲氧基)-苯基-(1,3.4) 噻二唑15.73g(0.076mol),加入甲苯磺酸 (10%),高速搅拌30 min,控制温度为80℃ ,TLC检测反应终点。将用冰浴冷却至0℃缓慢滴加 NaBH43.13g(0.0828mol),滴毕 ( 1h) ,保温反应 2h。冷却至室温,减压蒸馏除去溶剂[11-17],剩余物中加入乙酯乙酸288mL,滴加3mol/L盐酸116mL,分液,有机相依次用水 ( 2 ×142mL) 和饱和食盐水 (2 ×128 mL) 洗涤,无水硫酸镁干燥,通过重结晶 (石油醚:乙酯乙酸=5:1),得到目标化合物19.4g(0.055mo),总收率为75.8%,纯度为99.1%。
1HNMR(DMSO-d6,500MHz):9.78 (S,1H,OH),8.23 (t,J=5.5Hz,1H),7.66-7.69 (m,1H),7.25 (d,J=7.0Hz,1H),7.12 ( t,J=7.5Hz,1H,7.02-7.04 ( m,2H),6.82-6.86 (m,1H),6.79 (t,J=7.5HZ,1H),4.46 ( d,J=5.5Hz,2H,CH2),3.81 (s,3H,OCH3);
13CNMR (DMSO-d6),125MHz):168.42,161.76,157.36,156.68,129.51,127.97,128.35 >124.89,123.94,118.91,116.73,115.82,114.98,55.77,43.89;IR ( KBr):3447,3328,3081,3038,2963,2838,1609,1576,1465,1476,1345,1256,1249,1175,1108,1036,839,805,769,752。
2 结果与讨论
2.1 “一锅法” 合成影响因素
2.1.1 反应时间对收率的影响 时间短,反应不完全,时间过长,原料的自身缩合反应增加,反应液变黑,后处理困难,考虑收率和原子经济性,当反应时间4.5h 收率最佳 (表1)。
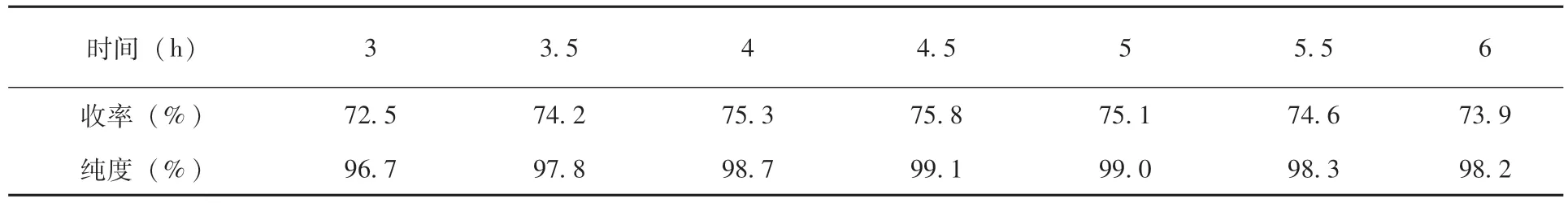
表1 反应时间对收率的影响
2.1.2 物料比对合成收率的影响 n1(邻异丙基水杨醛),n2(2-氨基-5-(4-甲氧基)-苯基-(1,3.4) 噻二唑),n3(NaBH4) 考察物料比对收率的影响,结果 (表2)。随着2-氨基-5-(4-甲氧基)-苯基-(1,3.4)噻二唑用量的增加,反应收率逐步提高。当n1:n2:n3=1:1.056:1.15 时,收 率 最 高 (75.8% ) (表 2) 。
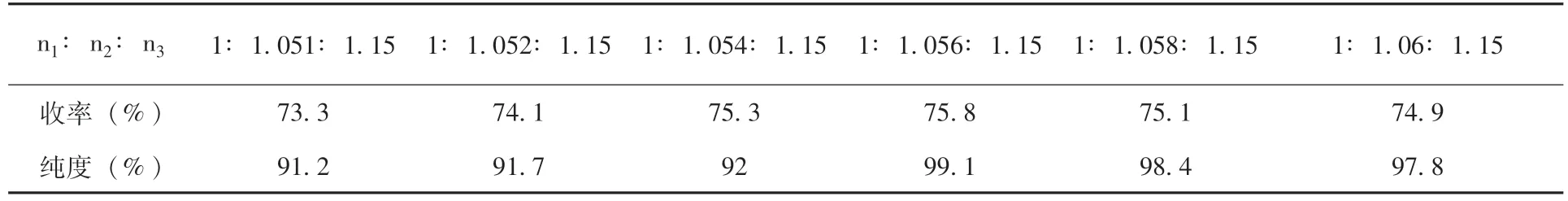
表2 物料比对合成收率的影响
2.1.3 不同溶剂对合成收率的影响 反应溶剂的选择对反应有很大的影响。选用不同溶剂条件下产品收率和纯度的数据 (表3)。在乙醇作为溶剂时反应收率最高,可达75.8%,并且副反应少,杂质含量少,目标产物的含量达99.1% (表3)。因此,使用乙醇作溶剂,收率最高 (表3)。

表3 不同溶剂对合成收率的影响
2.1.4 还原反应温度对收率的影响 反应温度是影响还原反应造择性的重要影响因素。从-5℃上升到0℃,收率随着温度的升高而增加,但是从0℃到30℃,收率反而随着温度的不断上升而减少,反应温度高于时,收率明显下降,随着温度升高,副反应加剧,造成收率下降,因此选择还原反应温度为0℃(表4)。

表4 温度对收率的影响
2.2 目标化合物波谱分析 1 450、1 500、1 560、1 600 cm-1左右的尖峰为苯环的骨架振动的吸收峰,以1 500 cm-1左右的吸收峰强度最大;1 250 cm-1左右的尖峰为C-O-C不对称伸缩振动的吸收峰。以上分析结果表明所合成的化合物与预期的结构相符。合成的2-[5-(4-甲氧基苯基)-(1,3.4)-噻二唑基-6-异丙基]胺基甲基苯酚,未见文献报道。产品经元素分析、IR、MS 和1H NMR表征,证明合成产物的结构与预期结构相符,经红外光谱、核磁共振氢谱1H NMR表征证明。
用傅里叶红外光谱仪测定产物的IR光谱。3 430、3 281 cm-1处 为 N-H的伸缩振动峰,1 624 cm-1处为羰基的伸缩振动峰,1 592cm-1处中强而尖的吸收峰为噻唑环的S—C=.的伸缩振动峰,1 507 cm-1处为仲酰胺N-H的弯曲振动峰,1 552 ~1 403 cm-1为苯环骨架振动特征峰,1 354 ~1 203 cm-1处为仲胺的C-N的伸缩振动峰。1H NMR表征证明合成产物为目标化合物。
3 结论
3.1 该合成技术工艺过程简单、生产安全、反应条件温和、生产成本降低、环境友好、产品收率高。
3.2 “一锅法” 合成工艺减少了溶剂的用量和废物排放量,收率较高。
猜你喜欢
能源化工(2021年3期)2021-12-31 11:59:23
烟台大学学报(自然科学与工程版)(2021年1期)2021-03-19 08:38:52
农药科学与管理(2019年8期)2019-11-23 08:04:44
兴义民族师范学院学报(2018年5期)2018-12-18 03:16:54
广州城市职业学院学报(2016年2期)2016-07-25 07:39:30
化工生产与技术(2016年5期)2016-03-13 10:07:26
中国塑料(2015年12期)2015-10-16 00:57:12
化学工业与工程(2015年1期)2015-02-10 03:01:33
郑州大学学报(理学版)(2014年4期)2014-03-01 04:21:19
无机化学学报(2014年12期)2014-02-28 17:34:01
