
含磷酸胍基间作用的磷酸双乙酸胍晶体电子结构与光学性质研究*
2019-04-10 02:45王磊涂兵田
物理学报 2019年6期
王磊 涂兵田
1) (西安石油大学,材料科学与工程学院,西安 710065)
2) (武汉理工大学,材料复合新技术国家重点实验室,武汉 430070)
(2018 年8 月31 日收到; 2019 年1 月15 日收到修改稿)
基于磷酸胍基间作用在L-精氨酸磷酸盐晶体特异性与生物化学功能中的重要角色,已制备了含有磷酸胍基的新晶体磷酸双乙酸胍. 本文采用第一性原理计算了磷酸双乙酸胍晶体的电子结构与三个晶向上的光学性质,探讨了其中基团间作用与光学性质的关联. 结果表明,磷酸双乙酸胍晶体能隙为4.77 eV,远小于磷酸二氢钾晶体,更易吸收光子,在胍基、羧基与磷酸根上发生电子跃迁. 磷酸双乙酸胍晶体在[100]和[010]方向光学性质相近,[001]晶向上胍基N-2p 在价带内电子跃迁产生强吸收,能量损失高且分布较窄,光学应用受到限制,该研究对理解和研究磷酸双乙酸胍晶体中基团间作用及其光学性质奠定了良好的基础.
1 引 言
在无脊椎动物体内,精氨酸磷酸(phosphate arginine,PA)作为主要的磷源和能量存储单元而广泛存在. PA 分子实现其生化功能的主要机制在于其分子中磷酸与精氨酸胍基间特殊的静电作用[1−4],除此之外,磷酸与胍基间特殊的非共价键作用在很多其他生物分子功能中也扮演着重要角色[5,6]. 在晶体中也存在类似的磷酸胍基. 20 世纪70 年代,Cotton 等[7]就以磷酸甲基胍、磷酸二甲基胍晶体结构中磷酸与胍基间氢键作用作为生物体中该类非共价键作用的模型,其研究中提到L-精氨酸磷酸盐(LAP)晶体结构中磷酸与胍基的关系更类似于生物分子的化学状态.
LAP 晶体是一种性能优异的非线性光学材料,其有效非线性光学系数约为磷酸二氢钾(KDP)晶体的2—3.5 倍,可以实现高达90%以上的转化效率,且具有超高的激光损伤阈值,其氘化晶体(DLAP)曾被认为是可以取代KDP 晶体用于激光惯性约束核聚变等领域的首选材料[8−10],因而有关该晶体的结构与性质,以及L-精氨酸衍生物晶体已有大量研究与报道[11,12]. 在探索LAP 晶体特异性的过程中,非生物环境下,磷酸溶液中L-精氨酸分子在不同能量下的构象转变被发现[13]; 磷酸与胍基间的作用会影响L-精氨酸分子聚集体产生的荧光发射现象也有报道[14]. 为了进一步研究磷酸胍基间的作用与晶体宏观性质机制的重要关联,已设计制备了含有类似磷酸与胍基的磷酸双乙酸胍(phosphate bis-guanidinoacetate,PBGA)晶体[15].
PBGA 晶体是属于三斜晶系,P-1 空间群,晶胞参数为:a=7.776(2) Å,b=8.113(2) Å,c=12.459(3) Å,α=89.591(2)°,β=89.146(3)°,γ=61.37°,V=689.8(3) Å3,D=1.599 g /cm3,Z=1. 图1为PBGA 晶体的分子构型及沿a轴的晶胞投影图,两个胍基乙酸与一个磷酸根阴离子组成了PBGA 分子,其中两个乙酸胍解离形式不同,一个的羧基完全失去质子形成COO-基团,另一个羧基则没有失去质子. 从投影图可以看出,沿晶体a轴方向的氢键连接形成了明显的层状结构.
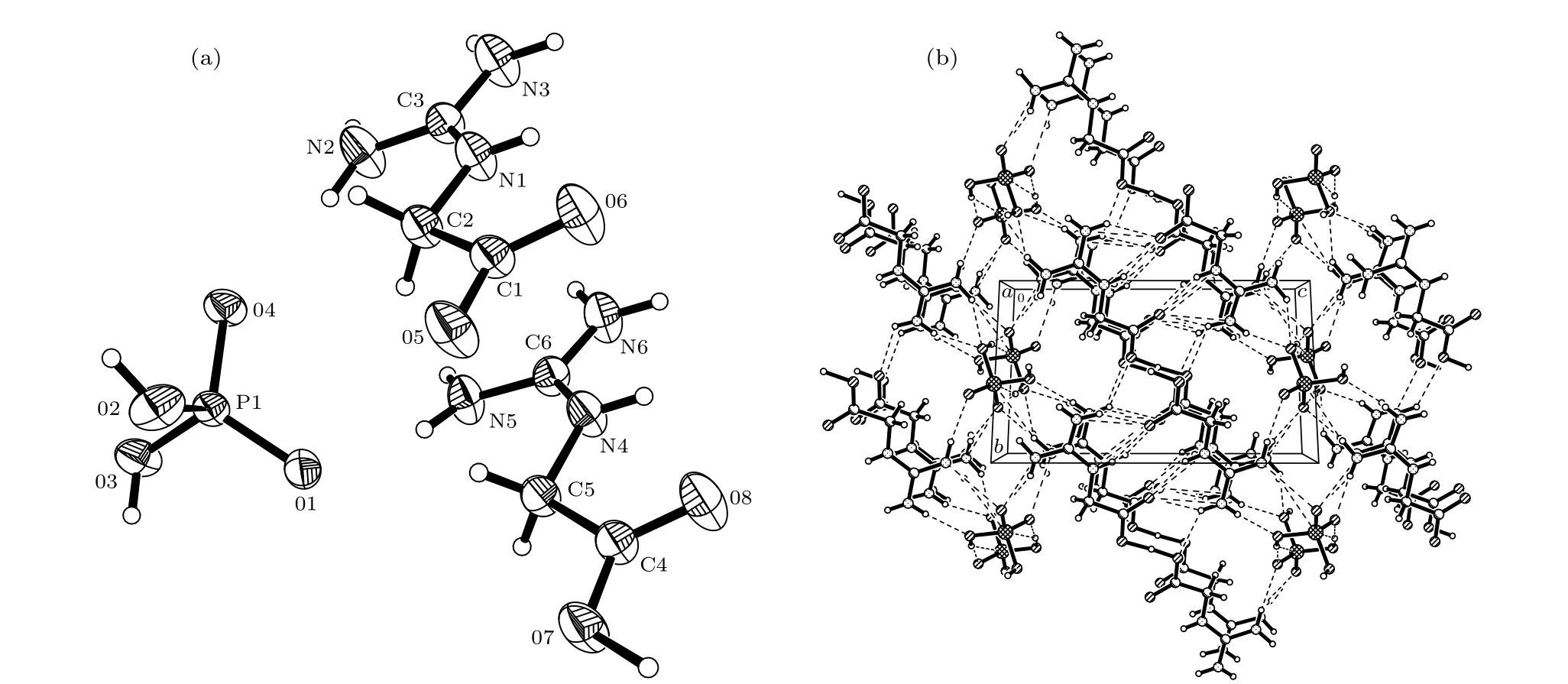
图1 PBGA 晶体的(a)分子构型和(b)沿a轴方向的投影图Fig. 1. (a) Molecular configuration and (b) projection viewed alonga-axis of PBGA crystal.
基于第一性原理在物质电子结构与宏观性质方面理论研究的可靠性,本文采用第一性原理的密度泛函理论(density functional theory,DFT)计算PBGA 晶体的能带结构与态密度,研究了该晶体在三个方向上的介电函数、折射率、吸收光谱、反射率及能量损失函数等光学性质,探讨晶体中基团间的作用与宏观光学性质的关联.
2 计算模型与方法
本文计算工作是在Materials Studio 8.0 软件中基于DFT 结合平面波赝势方法的CASTEP 软件包下进行的[16]. 采用实验所获PBGA 单晶结构,建立晶体模型如图2(a)所示,结构优化采用周期性边界条件,电子与电子之间的相互作用中的交换相关效应通过广义梯度近似(GGA)进行校正,采用超软赝势描述离子实与价电子之间的相互作用势,用GGA 中的Perdew-Burke-Emzerhof 处理电子之间的交换关联能[17−19]. 在倒易的k空间中,通过平面波截断能量(Ecut)的选择来控制平面波基矢的多少,从而改变计算精度. 通过收敛性测试确定了平面波截断能量Ecut=460 eV,自洽收敛精度设为2 × 10–5eV/atom,原子间的相互作用力收敛标准为0.05 eV/Å,原子的最大位移收敛标准为0.002 Å,晶体内应力不大于0.1 GPa,系统的总能量和电荷密度在布里渊区的积分计算使用Monkhorst-Pack 方案来选择k空间的网格点[20],布里渊区k矢的选取为9 × 9 × 9. 从图2(b)可以看出优化结果比较合理,电子结构与光学性质在同样设置下进行.
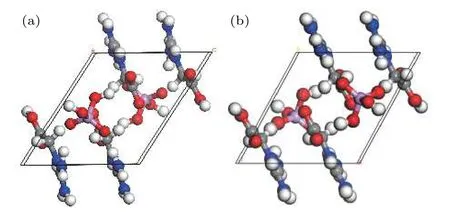
图2 PBGA 晶体(a)原始模型与(b)优化后模型Fig. 2. (a) Original and (b) optimized model of PBGA crystal.
材料的光学性质是物理性质中最重要的方面之一,在较小波矢下物质对光场的响应为线性,在此响应范围内固体宏观光学响应函数可用光的复介电常数ε(ω)=ε1(ω)+iε2(ω) 描述. 介电函数的实部ε1(ω)和虚部ε2(ω) 均由占据和未占用电子态之间的动量矩阵元获得,并由CASTEP 使用(1)和(2)式直接计算. 其他的光学性质如折射率n(ω)、 消光系数K(ω)、吸收系数I(ω) 、反射率R(ω) 、损失函数L(ω) 是根据(3)—(7)式得到[21,22]:
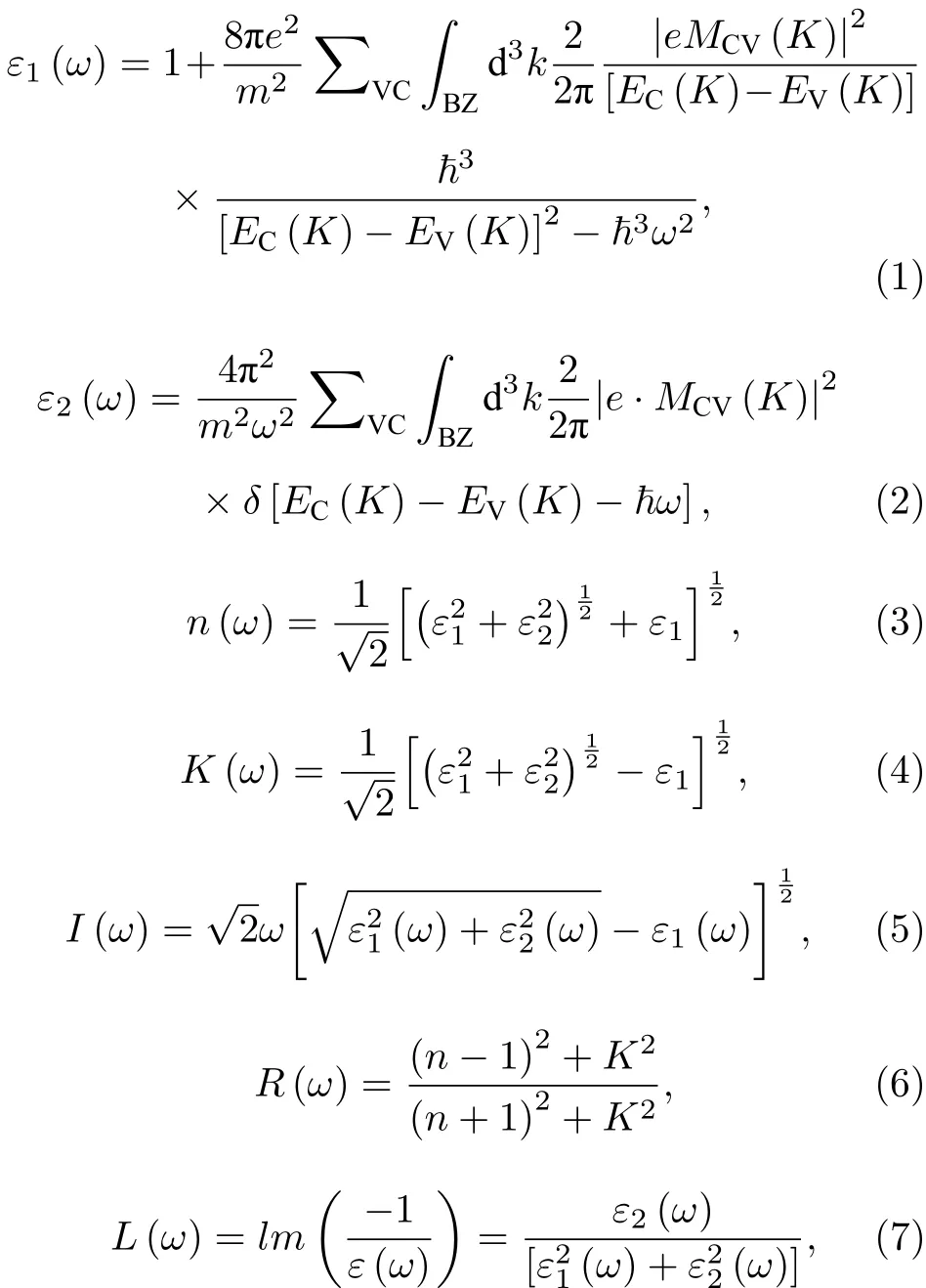
式中ω是光子频率;e是单元电荷; 下标C,V 分别表示导带和价带; BZ 为第一布里渊区;K为倒格矢;|MCV(K)|为动量矩阵元;EC(K)和EV(K)分别为导带和价带的本征能级.
3 结果与讨论
3.1 电子结构
图3(a)是PBGA 晶体的总态密度与轨道分态密度,根据轨道态密度对总态密度的不同贡献,价带可以分为如下三个区域: –15 eV 以下的低能级,–15 eV 至–9 eV 的中能级以及–9 eV 至0 eV 的高能级. 可以看出价带低能级主要是s 轨道的贡献,价带中能级是s 与p 轨道的综合贡献,价带高能级与导带中的底部主要由p 轨道组成,导带顶部主要来自s 轨道. PBGA 晶体的能带间隙为4.77 eV,比KDP 晶体的5.96 eV 小得多[23]. 根据电子跃迁理论,价带和导带之间的光子跃迁至少需要克服与带隙大小相同的能量窗. 因此,当355 nm 激光辐照晶体时,它可以吸收两个光子引起电子跃迁.PBGA 的窄带隙使得价带和导带之间的光子跃迁变得容易,对应光子的吸收相对较大.
图3(b)给出了总态密度与晶体中各原子分态密度,结合图3(a)可以看出,价带中低能级主要是由O-2s 和N-2s 贡献,–15 eV 至–5 eV 间能态由多个原子s 与p 轨道综合贡献,–5 eV 至0 eV 间能态主要来自于C-2p,O-2p 和N-2p,其中价带顶部主要由O-2p 和N-2p 组成,导带的底部主要来自C-2p.
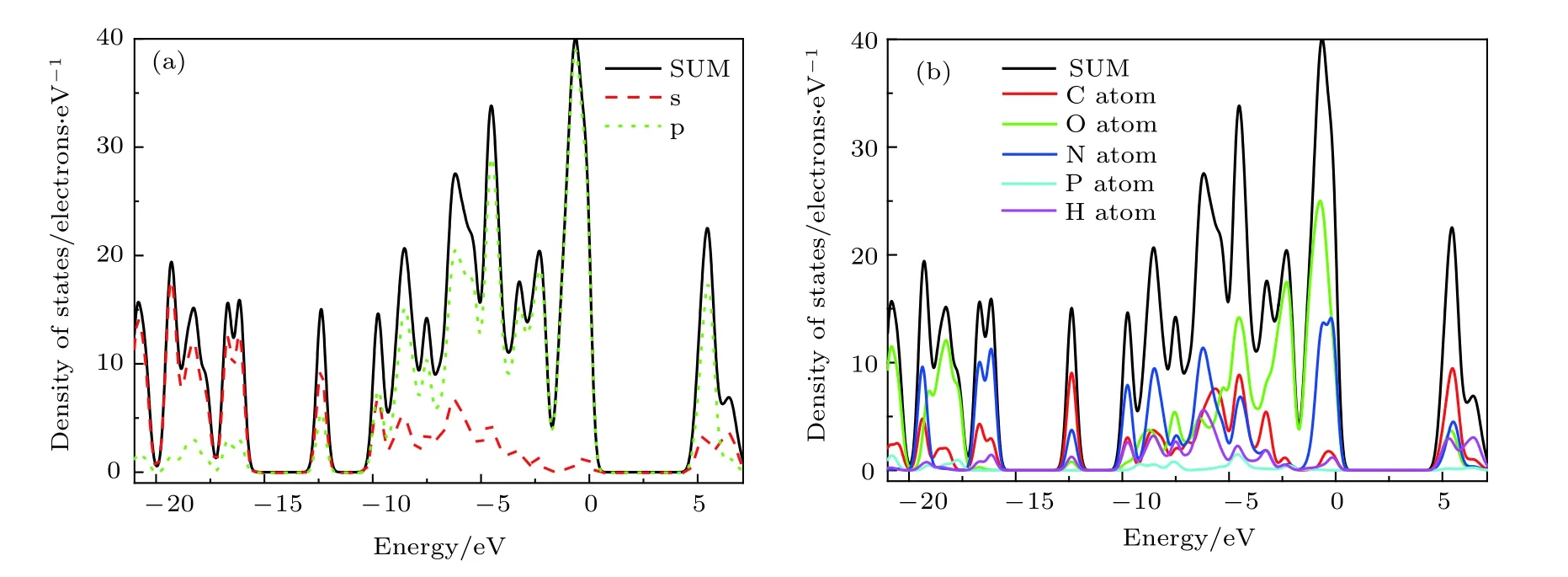
图3 PBGA 晶体的分态密度Fig. 3. Partial density of states of PBGA crystal.
图4 (a)为O-2p 总态密度与不同基团上O-2p态密度分布,图4(b)为N-2p 总态密度与不同基团上N-2p 态密度分布,可以看出价带顶部电子能态主要来自于乙酸胍分子胍基上N-2p,磷酸根与羧基上O-2p 的贡献,其中带电乙酸胍分子相对不带电乙酸胍分子,其O-2p 占据比例更高. 因此,当光子入射进入PBGA 晶体时,胍基、磷酸根与羧基上最容易发生电子跃迁.
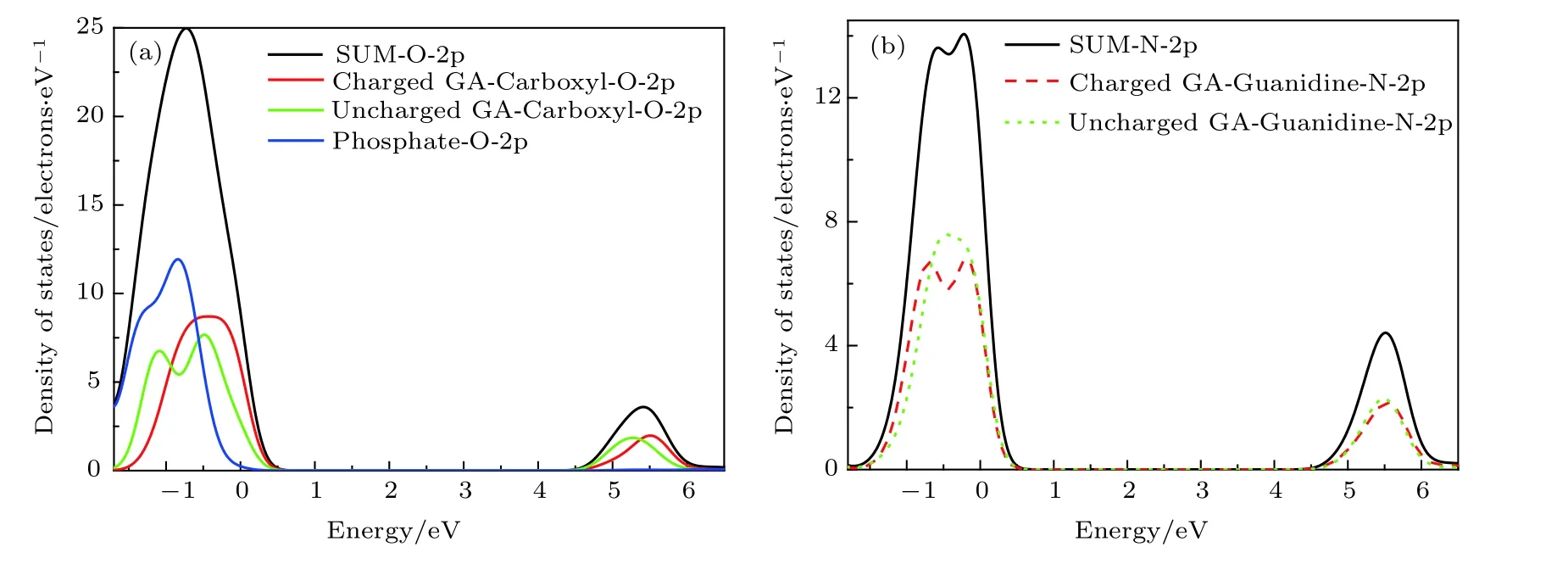
图4 PBGA 晶体中(a)氧和(b)氮的p 轨道分态密度Fig. 4. The p-orbital partial density of states of (a) oxygen and (b) nitrogen in PBGA crystal.
3.2 光学性质
PBGA 晶体在[100],[010]以及[001]三个方向介电函数虚部与能量的关系曲线如图5 所示. 从图5 可以看出,PBGA 晶体在三个方向的首个介电峰分别位于6.22,5.78 和6.07 eV,且在[001]方向强度最强,[100]方向最弱. 结合态密度图3 可知,首个特征峰主要是由PBGA 中N-2p 与O-2p态由价带顶部跃迁至导带引起.
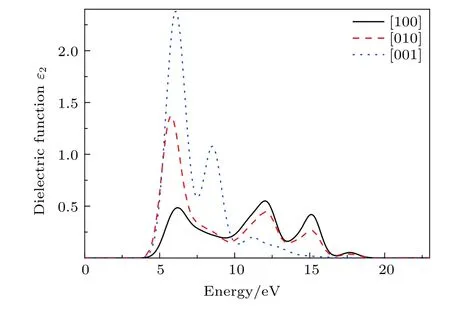
图5 PBGA 晶体的介电函数虚部与能量关系Fig. 5. Relationship between energy and imaginary part of dielectric function of PBGA crystal.
同时可以看出,PBGA 晶体在[100]和[010]两个晶向上除了首个介电峰有大小差别外,这两个方向上对大于7.50 eV 的光子能量响应基本一致,在12.01 和15.08 eV 都分别出现了两个较明显的介电峰. 结合态密度分析,这两处介电峰应该分别来自于O-2p 上电子自–7 eV 左右与N-2p 上电子自–10 eV 左右跃迁至导带C-2p 产生. 而[001]方向上位于8.53 eV 的强峰和11.15 eV 的弱峰,相较于其他两个方向谱峰明显向低能量偏移,其中8.53 eV 处介电峰响应强度明显强于其他两个方向,结合态密度分析,在光子能量小于10 eV 时,该方向上介电峰应该来自于胍基上N-2p 自价带中部 –8.50 eV 左 右 处 到 价 带 顶 部 的 跃 迁,表 明PBGA 晶体在[001]晶向上电子跃迁时存在此类特殊的中间能级. 随入射光子能量增强,该类跃迁减弱且不再发生,可以看出由H-1s 与N-2p 自–6.20 eV 处至导带的电子跃迁,在11.15 eV 处产生了较弱的介电峰.
图6(a)为PBGA 晶体在三个不同晶向[100],[010]和[001]的复折射率曲线. 入射光子能量小于3.70 eV 时,消光系数K都为0,而折射率n初始值分别为1.12,1.18 和1.25 且随光子能量增加变化不明显,对应于其在该区域吸收很弱. 随光子能量增加,n和K都先增后减,峰值分别位于5.30 和6.30 eV 左右. 当光子能量大于19 eV 时,K值减小为0,n近似成为一个固定值,复折射率几乎为常数.
PBGA 晶体[100],[010]和[001]晶向的吸收系数变化曲线如图6(b)所示,总体吸收范围3.70—9.30 eV,根据λ=ch/Ek=1240/E,晶体的吸收截止波长在335 nm,表明其在可见光范围也具有良好的透过性能.
三个晶向上的第一个吸收峰分别位于6.46,6.05 和6.43 eV,[001]方向8.82 eV 处出现了其他两个方向没有的明显吸收峰,强度稍弱于其首个吸收峰,与该方向介电峰的电子跃迁对应,当光子能量大于10 eV,三个方向上的吸收强度发生反转,[100]方向强度最高,[010]方向次之,这两个晶向上12.22 和15.24 eV 处都分别出现了两个较强的吸收峰,对应于O-2p 与N-2p 自价带中部向导带的跃迁. 由于8.82 eV 处强吸收来自于价带内胍基上N-2p 从中部到顶部的跃迁,使得电子离域性降低,导致当入射光子能量增大后,[001]方向跃迁电子较少,吸收最弱,且在光子能量约为17.20 eV时,吸收较其他两个方向提前截止.

图6 PBGA 晶体的(a)折射率和(b)吸收系数Fig. 6. (a) Refractive index and (b) absorption coefficient of PBGA crystal.
图7 为PBGA 晶体的反射率和能量损失函数图谱. 从图7(a)可以看出,晶体三个方向反射峰的极值分别出现在5.81,5.98 和6.55 eV 处,与介电函数虚部曲线趋势类似,[001]方向上在9.23 eV处出现了第二个强反射峰,迅速下降到与其他两个方向反射相当的强度,该处[010]和[100]两个方向的反射率非常低,且差别不大,当光子能量高于9 eV 后,这两个晶向在12.43 和15.53 eV 都出现了两个较明显的反射峰. 在17 eV 以上,三个方向的反射率趋于一致,23 eV 以上降为0. 反射率分析表明该晶体三个晶向上对光子反射性质有明显差异,相对于[010]和[100]两个方向较宽且均匀的反射性质,[001]方向上的反射率分布较窄且集中在低能量区,该现象对于该晶体的光学器件开发与应用具有一定的指导意义.

图7 PBGA 晶体的(a)反射率与(b)能量损失函数Fig. 7. (a) Reflectivity and (b) loss function of PBGA crystal.
能量损失曲线是材料对光子吸收与反射的综合效果. 从图7(b)可以看出,PBGA 晶体在光子能量为3.70 eV 时开始有能量损失,其中[100]和[010]两个方向的能量损失曲线相似,分布较宽,这两个方向分别在6.58 和6.62 eV 时出现第一个能量损失峰,对应于其反射性质与分子上电子跃迁产生的吸收性质. 随光子能量增大,能量损失降低后再次增大,这两个方向分别在12.55 和15.50 eV 出现了两个强于[001]方向的损失峰,与介电峰对应,是反射与吸收的加和. 在光子能量高于23 eV 后,能量损失系数降为0,光子能量不再被吸收或反射,能够全部穿透材料. 与反射率曲线类似,[001]方向对光子能量的损失曲线分布较窄,7.15 和9.46 eV出现的两个损失峰强度最高且尖锐,随后较弱的第三能量损失峰出现在11.27 eV,在光子能量高于18 eV 后,能量损失系数降为0.
综合以上PBGA 晶体的光学性质可以看出,晶体在[100]和[010]方向的光学性质相近,其吸收全部来自于电子自价带到导带的跃迁,而[001]晶向中会产生价带内部胍基上N-2p 的电子跃迁,且反射率分布较窄并明显高于其他两个方向,结合能量损失曲线,该晶体在[001]晶向的光学性质会限制其在光子能量较低时的光学应用.
4 结 论
本文采用基于密度泛函理论的第一性原理平面波超软赝势方法,对含有磷酸与胍基的PBGA晶体进行了电子结构与光学性质的理论研究. 结果表明,PBGA 晶体能隙为4.77 eV,比KDP 晶体更易于吸收光子,在磷酸根、胍基和羧基三个基团上发生电子跃迁. PBGA 晶体在[001]晶向上存在胍基N-2p 价带内电子跃迁产生的强吸收,使该晶向光学性质异于其他两个方向,此性质对该晶体的器件开发与光学应用有一定指导意义. 下一步研究将关注价带内胍基上电子跃迁是否与磷酸基团相关,目前的结果对理解和研究PBGA 晶体中基团间作用及其光学性质具有一定意义.
猜你喜欢
魅力中国(2021年17期)2021-11-26
人工晶体学报(2021年10期)2021-11-26
磷肥与复肥(2021年8期)2021-09-28
铁道建筑技术(2021年4期)2021-07-21
有色设备(2021年4期)2021-03-16
发明与创新·大科技(2019年11期)2019-03-07
百科知识(2017年3期)2017-03-17
中国美容医学(2004年3期)2004-09-17
