
基于高通量测序研究草莓根际微生物群落结构和多样性①
2019-04-08 09:13赵密珍庞夫花
土壤 2019年1期
赵 帆, 赵密珍, 王 钰, 关 玲, 庞夫花
基于高通量测序研究草莓根际微生物群落结构和多样性①
赵 帆1,2, 赵密珍1*, 王 钰2, 关 玲1, 庞夫花1
(1 江苏省农业科学院果树研究所/江苏省高效园艺作物遗传改良重点实验室,南京 210014;2 安徽大学资源与环境工程学院,合肥 230601)
研究草莓根际土壤微生物群落组成和结构,对健康草莓土壤生态系统的构建和保持具有重要意义。以不同地区草莓根际土壤为研究样本,利用Miseq平台Illumina第二代高通量测序技术并结合相关生物信息学分析土壤细菌16S rRNA基因V4+V5区域和真菌ITS1+ITS2区域的丰富度和多样性指数以及群落结构。结果表明:从15个草莓根际土壤样本中获得4 554个细菌分类操作单元OTU和1 298个真菌OTU,草莓根际土壤的优势细菌门为变形菌门、厚壁菌门、放线菌门、酸杆菌门和绿弯菌门,主要的优势细菌属有16种;优势真菌门为子囊菌门、接合菌门和担子菌门,主要的优势真菌属有8种。冗余分析(RDA)显示,全氮和pH对土壤微生物群落结构的影响最大,共解释了61% 的群落变化,各因子的贡献率大小依次为土壤全氮>pH>有效磷>全钾>全磷>有机质>速效钾>碱解氮;相关性分析也表明,土壤理化指标均与不同优势菌门存在密切的相关关系。本研究结果加深了对草莓根际微生物群落结构和多样性的认识,为深入研究草莓根际微生物多样性及功能与环境因子之间的关系提供了借鉴。
草莓;根际土壤;Illumina高通量测序;土壤微生物群落;土壤理化性质
草莓(Duch.)属蔷薇科(Rosaceae)草莓属植物,是一种营养价值和经济价值较高的水果,因其色、香、味俱佳,深受消费者青睐。草莓在同一地块上连续种植两年以上,地下病虫害逐年加重,草莓长势逐年变弱,表现为植株发育受阻,甚至死亡;果品质量逐年变劣,产量逐年下降。特别是近年来设施农业的快速发展,设施草莓栽培面积逐渐增加,草莓连作障碍发生越来越严重[1],严重制约了草莓产业的发展,亟待解决。
土壤根际环境是一个特殊的生态系统,它与非根际土壤在生物学、化学等特性上具有显著差异[2]。土壤根际环境受植物根系的影响,是有益微生物及有害微生物同宿主植物发生相互作用的一个区域,蕴含着充足的微生物资源[3]。土壤微生物能促进土壤有机质的分解及土壤养分的转化,对维持土壤质量及推动土壤的健康发展具有十分重要的作用[4]。土壤微生物能够影响土壤养分的吸收和转化,微生物群落结构的失衡是连作障碍的主要原因[5]。
传统的土壤微生物研究方法如微生物平板培养法、生物标记法、Biolog鉴定系统法等[6]会过低估计土壤微生物的群落结构组成,很难详细描述出土壤微生物的群落结构组成方面的信息,也无法描述不同群体的生理差异。第二代测序Illumina MiSeq方法有效避免了通量低、操作复杂和准确率低等缺陷[6-7],具有操作简单和成本较低的优势,并采用边合成边测序原理,结果可信度高。MiSeq高通量测序平台,不仅实现了对多样品的多个可变区同时测序,而且在测序速度和测序通量上都进一步得到提升。目前此平台在微生物多样性群落结构研究方面得到了广泛的应用[8-9]。
一些研究发现随着草莓连作程度的加深,根际土壤中细菌数量逐渐降低,真菌数量逐渐增加,细菌和真菌的比值逐渐降低,土壤微生物由“细菌型”向“真菌型”转变,说明土壤真菌与草莓连作的关系密切,其中病原真菌是草莓连作障碍因子之一,主要包括镰刀菌属、轮枝菌属、丝核菌属、腐霉属、疫霉属、拟盘多毛孢属和炭疽菌属致病真菌。国内研究结果表明,草莓病原真菌以镰刀菌和丝核菌为主[10]。草莓根际土壤微生物群落组成和结构目前还不清楚,本研究以不同地区草莓根际土壤为材料,采用高通量测序技术,探讨不同地区草莓根际土壤微生物群落组成和结构,以期为草莓根际微生态环境研究提供理论依据。
1 材料与方法
试验于2016年9—11月在江苏省农业科学院草莓温室大棚进行。
1.1 试验材料
试材为生长整齐一致的三叶一心期“紫金久红”草莓苗。盆栽土壤分别采集于江苏省4个地区草莓连作大棚0 ~ 20 cm土层,分别是溧水白马(BM)、邳州港上(G)、东海石榴(S)、溧水傅家边(FJB),再从溧水白马一个多年没有开垦的农田耕作层取土盆栽作为对照(CK)。
1.2 试验设计
将不同土壤与珍珠岩以3∶1的体积充分混匀装盆。塑料盆直径16 cm,深20 cm,每盆装土1 500 g,每盆栽草莓1株,每种土壤栽种20盆。草莓苗于2016年9月10日定植,定植后早晚各浇一次水且适当遮阴7 d,之后正常水、肥、光照管理,温度维持在20 ~ 25 ℃,光周期约12 h (昼)/12 h (夜)。定植后50 d时(开花前)土壤取样及测定分析。分析用土壤来自长势较为一致的草莓根际土,采用抖土法获取粘附在3株草莓根系表面上的根际土壤并充分混匀作为1个土样,重复3次。所获取的土壤一部分保存于−20 ℃冰箱用于土壤微生物分析,其余根际土壤风干后过 100 目网筛用于土壤理化性质的分析。
1.3 试验方法
1.3.1 土壤理化性质测定 采用硫酸消解,微量凯氏定氮法测定土壤全氮含量;采用氢氧化钠熔融,硫酸钼锑抗比色法测定土壤全磷含量;采用火焰光度计法测定土壤全钾含量;采用碱解扩散法测定土壤碱解氮含量;采用重铬酸钾容量法-稀释热法测定土壤有机质含量;采用碳酸氢钠浸提,钼锑抗比色法测定有效磷含量;采用乙酸铵浸提,火焰光度计法测定速效钾含量;土壤pH用无CO2蒸馏水(水土质量比5∶1)浸提,pH计( METTLER TOLEDO)测定[11]。
1.3.2 土壤微生物基因组DNA的提取 提取试剂盒为Omega土壤微生物DNA提取试剂盒,土壤样品0.5 g,按试剂盒的试验步骤进行土壤微生物总DNA的提取,DNA样品于−20 ℃保存待用。
1.3.3 PCR扩增及Illumina高通量测序 提取样品总DNA后,根据设计得到细菌V4+V5 (F:5′-GTGCCAGCMGCCGCGG-3′,R:5′-CCGTCAATTCMTTTRAGTTT-3′)和真菌ITS1+ITS2 (F:5′-CTTGGTCATTTAGAGGAAGTAA-3′,R:5′-GCTGCGTTCTTCATCGATGC-3′)合成的引物,合并引物接头,进行PCR扩增。PCR反应体系:含15 mmol/L MgCl2的10×Buffer 10.0 μl,2.5 mmol/L dNTPs 2.0 μl,10 μmol/L引物各5.0 μl,5 U/μl Taq酶1.0 μl,DNA模板4.0 μl,灭菌去离子水73.0 μl,总体积100 μl。PCR反应条件:94℃ 5 min,94℃30 s,55℃ 30 s,72℃ 30 s,30个循环;72℃ 10 min。PCR扩增后,对其产物进行纯化、定量和均一化形成测序文库,建好的文库先进行文库质检,质检合格的文库用Illumina HiSeq PE250进行测序,测序由南京集思慧远生物科技有限公司完成。
1.4 生物信息分析方法
1.4.1 数据过滤及质量评估 数据过滤主要步骤:①去除平均质量值低于20的长Reads;②去除Reads含N的碱基数超过3个的长Reads;③Reads长度范围为220 ~ 500 nt。
1.4.2 序列优化 利用Mothur软件(version 1.34.4,http://www. mothur.org/),进行序列优化,主要步骤[12]为:①对序列进行筛选,去除模糊碱基数大于0、单碱基高重复区大于8、重叠区错配数大于0及长度大于97.5% 的序列(细菌)及重叠区长小于20 bp的序列(真菌);②进行去冗余处理。
1.4.3 OTU聚类分析 为便于下游物种多样性分析,将Tags聚类为OTU (operational taxonomic units)。首先,将拼接的Tags中的Singletons (对应Reads只有一条的序列)过滤掉,因为Singletons可能由于测序错误造成,故将这部分序列去除,不加入聚类分析。进而,利用Usearch在0.97相似度下进行聚类,对聚类后的序列进行嵌合体过滤后,得到用于物种分类的OTU,每个OTU被认为可代表一个物种。
1.5 数据分析
随机选取相似度在97% 条件下的OTU生成稀释曲线,并利用软件Mothur计算丰富度指数Chao和Observed species指数,多样性指数Simpson和Shannon。基于RDP和UNITE分类学数据库对OTU进行物种注释,并用Excel和SPSS进行数据处理,利用Excel和R语言工具对样品物种组成及相对丰度统计结果绘制柱状图和Veen图。采用Canoco软件进行冗余分析。
2 结果与分析
2.1 土壤理化性质
由表1可知,不同样品土壤理化性质表现出显著差异。其中BM土壤全氮、全磷、有机质、速效钾和有效磷含量显著高于其他样品,S土壤全钾和碱解氮含量显著高于其他样品,FJB 土壤pH偏低,属于酸性。
2.2 土壤样品测序深度评估
15个细菌样品测序,双端reads拼接后共产生623 498条Raw Tags,序列优化后共得到566 667条Clean Tags,聚类后一共获得4 554个OTU;15个真菌样品测序,双端reads拼接后共产生547 484条Raw Tags,序列优化后共得到542 038条Clean Tags,聚类后一共获得1 298个OTU。参照文献[13]随机抽取测序序列,将抽到的序列数与它们所能代表OTU的数目构建曲线,在97% 相似性水平下聚类OTU并制作各样品的稀释曲线(图1),可知细菌和真菌曲线逐渐趋向平坦,说明测序数量合理。
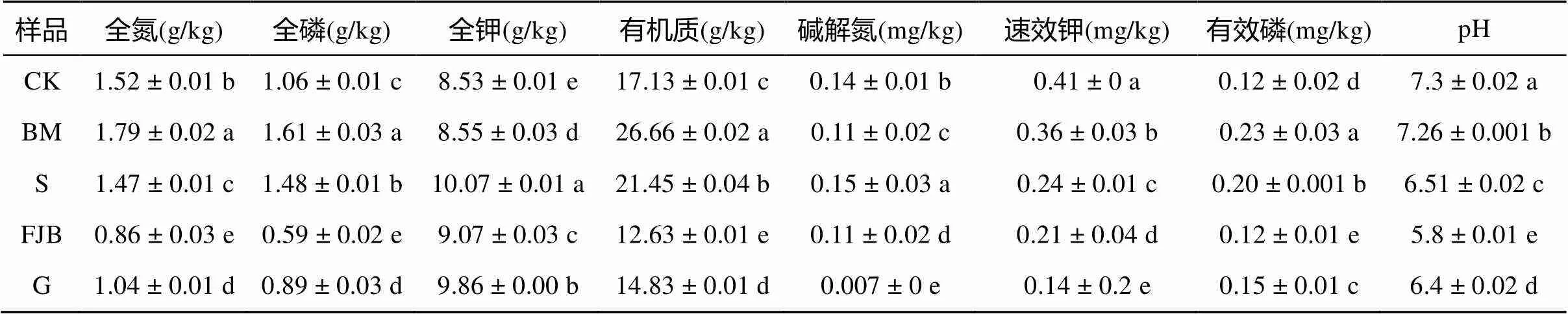
表1 草莓根际土壤理化性质
注:CK表示对照土壤,BM、S、FJB、G分别表示江苏溧水白马、东海石榴、溧水傅家边、邳州港上草莓连作土壤;同列不同小写字母表示不同土壤样品间差异在<0.05水平显著;下同。
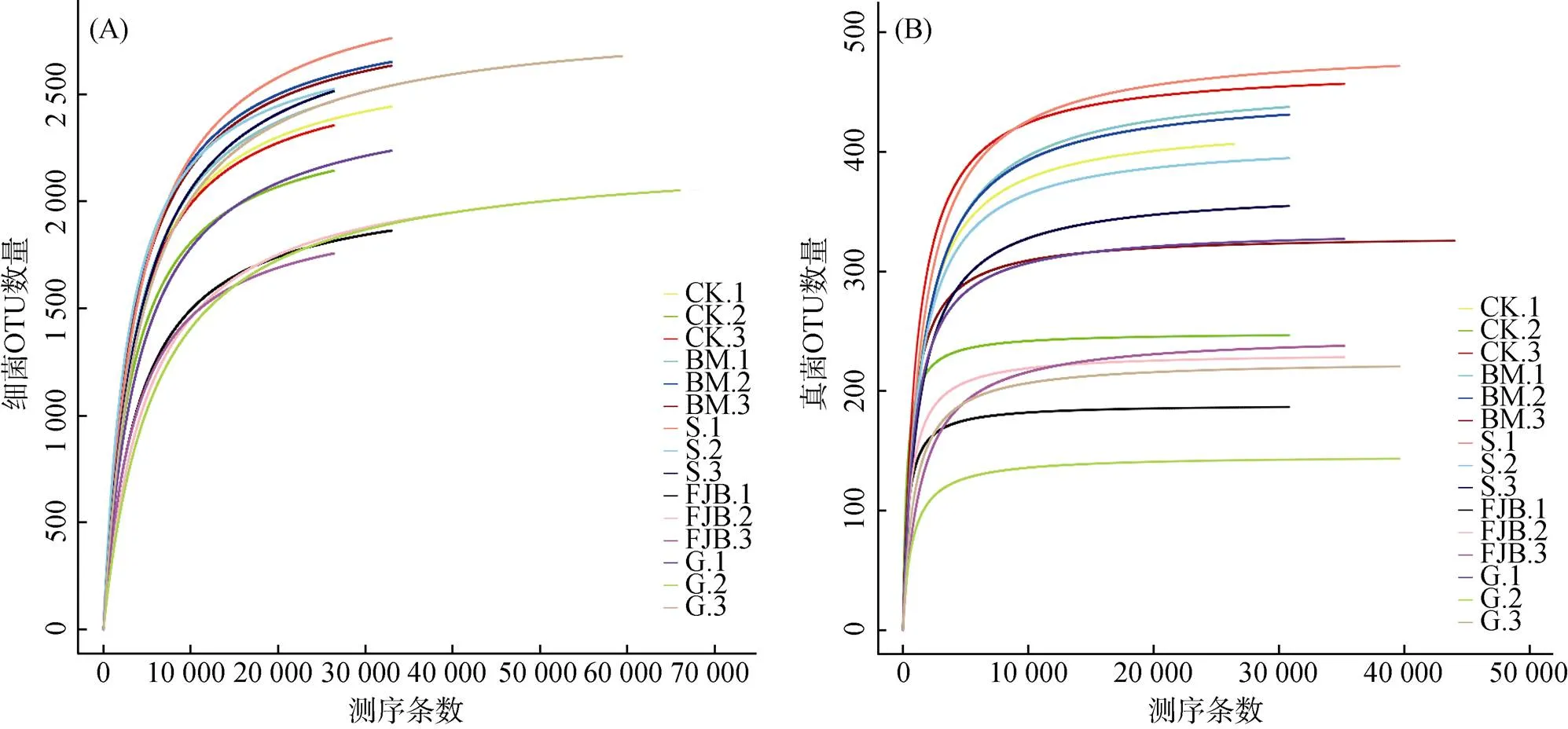
图1 土壤样品细菌(A)、真菌(B)多样品稀释曲线
2.3 草莓根际土壤微生物群落丰富度和多样性变化
Observed species指数和Chao指数可反映群落物种丰富度,由表2可知,BM以及S土壤细菌Observed species指数和Chao指数较高,相较于CK分别增加了10.80% 和14.50% 以及10.84% 和13.28%,但与CK无显著性差异;FJB以及G土壤细菌Observed species指数相较于CK分别减少了24.04% 和21.61%,均有显著性差异;Chao指数FJB与CK有显著性差异,G与CK差异不显著,BM、S和G分别与FJB有显著性差异。BM以及S土壤真菌Observed species指数和Chao指数相较于CK分别增加了4.18% 和7.06% 以及7.41% 和8.35%,但与CK无显著性差异;而FJB以及G土壤真菌Observed species指数和Chao指数较低,真菌Observed species指数相较于CK分别减少了40.74% 和40.88%,且有显著性差异,真菌Chao指数相较于CK分别减少了37.32% 和38.51%,FJB与CK有显著性差异,G与CK 无显著性差异,BM和S分别与FJB和G有显著性差异,FJB与G无显著性差异。由此可见,土壤细菌和真菌群落物种丰富度的变化因区域的不同而存在较大差异。
Simpson指数和Shannon指数可反映群落物种多样性。由表2可知,BM以及S土壤细菌Simpson指数和Shannon指数较高,相较于CK分别增加了3.12% 和5.34% 以及3.12% 和8.25%;FJB以及G土壤细菌Simpson指数和Shannon指数较低,FJB相较于CK其Simpson指数增加了1.04%,Shannon指数下降8.62%,G土壤相较于CK分别下降3.13% 和12.50%,但FJB、G与CK均无显著性差异。BM土壤真菌Simpson指数最高,相较于CK增加了2.13%,S、FJB和G土壤真菌Simpson指数都低于CK,分别减少了4.26% 和3.19% 以及32.98%,S、FJB与CK 无显著性差异,而G与CK 有显著性差异;土壤真菌Shannon指数BM、S、FJB和G都低于CK,分别减少了1.72%、9.78%、17.67% 和42.53%,除G与CK有显著性差异外,其他与CK 无显著性差异,BM、S、FJB之间无显著性差异,BM、S与G有显著性差异。由此,可见土壤细菌群落物种多样性的变化在不同区域间差异不显著,而真菌群落物种多样性存在差异。
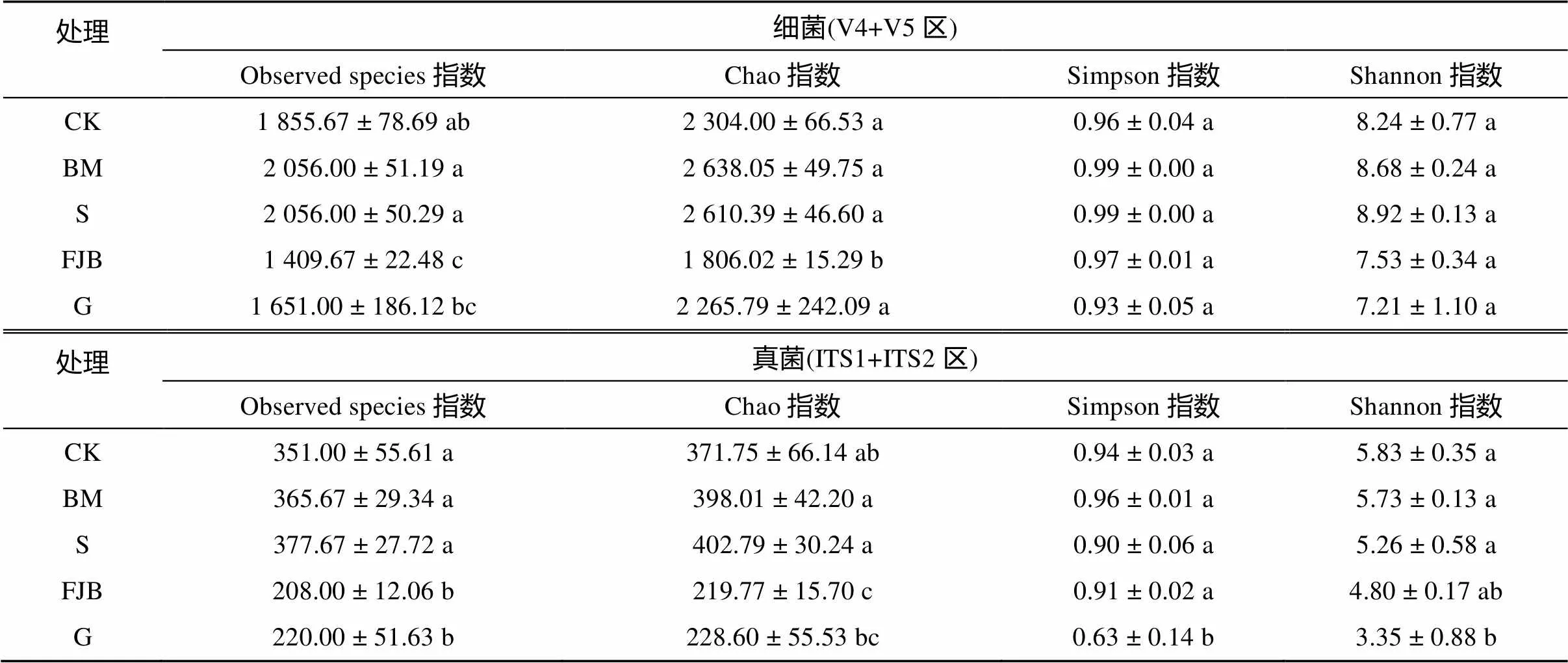
表2 草莓根际土壤细菌和真菌群落丰富度和多样性指数
2.4 草莓根际土壤微生物群落门类组成
从门的分类水平看,草莓根际土壤样本中主要检测出细菌10个门,分属变形菌门、厚壁菌门、放线菌门、酸杆菌门、绿弯菌门、芽单胞菌门、拟杆菌门、疣微菌门、蓝藻门、浮霉菌门。其中,变形菌门、厚壁菌门、放线菌门、酸杆菌门和绿弯菌门为草莓根际土壤的优势菌种(表3),草莓根际土壤样本中变形菌门的相对丰度为23.51% ~ 35.46%,厚壁菌门的相对丰度为11.51% ~ 29.17%,放线菌门的相对丰度为8.48% ~ 16.48%,酸杆菌门的相对丰度为6.4% ~ 15.14%,绿弯菌门的相对丰度为5.88% ~ 10.39%。在优势菌种中,BM、S和FJB土壤放线菌门的相对丰度高于G和CK土壤,且FJB与CK差异显著,BM、S、FJB和G土壤酸杆菌门的相对丰度都低于CK,且FJB与CK差异显著。由此可见,草莓连作后土壤中细菌群落组成发生了变化。
测序结果表明,从草莓根际土壤中鉴定得到的真菌主要来自5个门,包括子囊菌门、接合菌门、担子菌门、壶菌门和球囊菌门 (表3)。其中,子囊菌门、接合菌门和担子菌门为草莓根际土壤的优势菌种(表3),草莓根际土壤样本中子囊菌门的相对丰度为52.39% ~ 87.67%,接合菌门的相对丰度为1.88% ~ 7.79%,担子菌门的相对丰度为3.06% ~ 18.23%。子囊菌门相对丰度最高的为FJB(87.67%),依次为G(85.02%)、S(66.79%)、CK(60.35%)、BM(52.39%),担子菌门的相对丰度BM、S、FJB和G都小于CK。说明草莓连作后土壤中真菌群落组成发生了变化。
在属水平上,不同土壤样品细菌优势类群差异明显。其中CK土壤优势类群为乳球菌属(1.74%)、芽孢杆菌属(12.60%)和(1.11%);BM土壤优势类群为(3.44%)、中慢生根瘤菌属(1.71%)、芽孢杆菌属(6.58%)、乳杆菌属(2.19%)、链霉菌属(2.23%)、(2.81%)和丰佑菌属(1.29%);S土壤优势类群为芽孢杆菌属(7.04%)、链霉菌属 (3.21%)、(1.36%)、(1.04%)、芽单胞菌属(2.1%)和丰佑菌属 (1.14%);FJB土壤优势类群为伯克氏菌属 (7.68%)、(1.83%)、芽孢杆菌属(11.78%)、乳球菌属(2.44%)、链霉菌属(5.60%)、(1.29%)和芽单胞菌属(2.24%);G土壤优势类群为伯克氏菌属(1.28%)、芽孢杆菌属(19.65%)、乳球菌属(3.29%)、(2.68%)和链霉菌属(1.53%)。
对已确定分类地位至属的真菌分析表明,不同土壤样品真菌优势类群差异也明显(表3)。其中CK土壤优势类群为被孢霉属(5.25%);BM土壤优势类群为青霉菌属(6.8%)、曲霉属(4.45%)、(6.13%)、(3.62%)、被孢霉属(7.26%)和(2.72%);S土壤优势类群为青霉菌属(18.04%)、曲霉属(2.36%)、镰刀菌属(1.85%)、被孢霉属(7.41%)和鬼伞属(1.5%);FJB土壤优势类群为青霉菌属(28.52%)、曲霉属(13.87%)、支顶孢属(1.47%)、镰刀菌属(1.1%)和被孢霉属(2.63%);G土壤优势类群为青霉菌属(1.86%)、曲霉属(2.16%)、(1.44%)和支顶孢属(1.18%)。
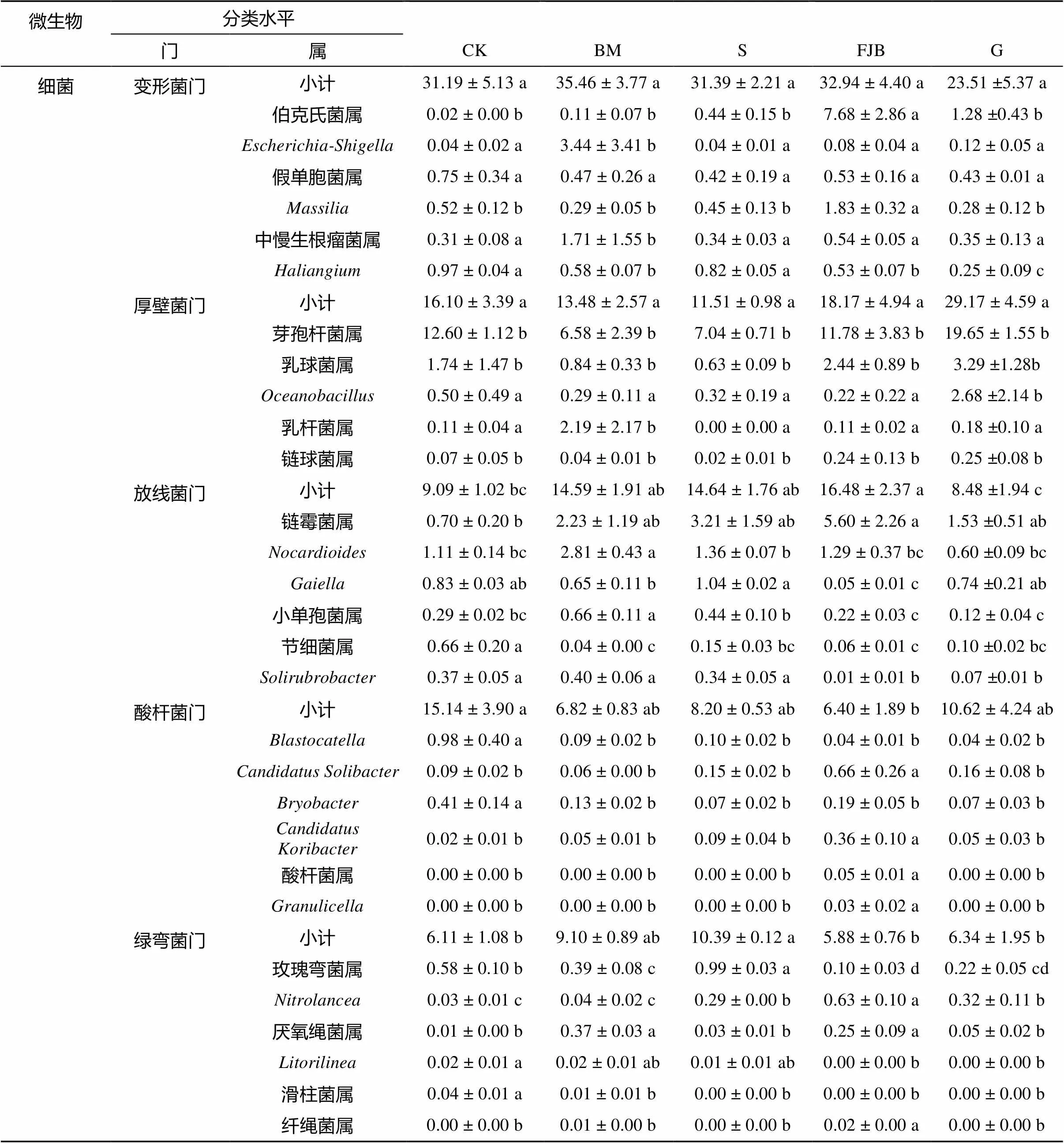
表3 草莓根际土壤微生物在门和属水平上的丰度变化(%)
续表
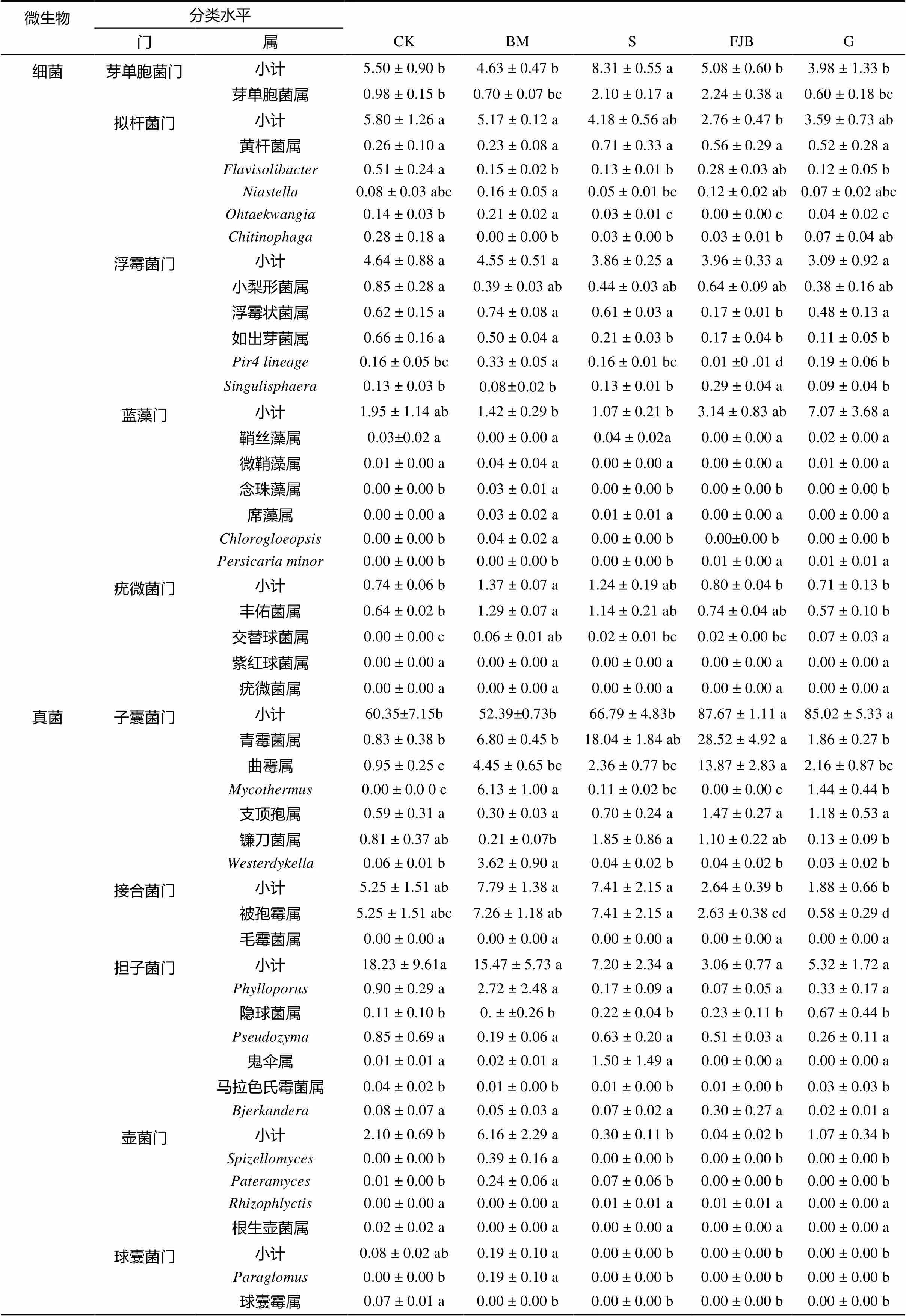
微生物分类水平 门属CKBMSFJBG 细菌芽单胞菌门小计5.50 ± 0.90 b4.63 ± 0.47 b8.31 ± 0.55 a5.08 ± 0.60 b3.98 ± 1.33 b 芽单胞菌属0.98 ± 0.15 b0.70 ± 0.07 bc2.10 ± 0.17 a2.24 ± 0.38 a0.60 ± 0.18 bc 拟杆菌门小计5.80 ± 1.26 a5.17 ± 0.12 a4.18 ± 0.56 ab2.76 ± 0.47 b3.59 ± 0.73 ab 黄杆菌属0.26 ± 0.10 a0.23 ± 0.08 a0.71 ± 0.33 a0.56 ± 0.29 a0.52 ± 0.28 a Flavisolibacter0.51 ± 0.24 a0.15 ± 0.02 b0.13 ± 0.01 b0.28 ± 0.03 ab0.12 ± 0.05 b Niastella0.08 ± 0.03 abc0.16 ± 0.05 a0.05 ± 0.01 bc0.12 ± 0.02 ab0.07 ± 0.02 abc Ohtaekwangia0.14 ± 0.03 b0.21 ± 0.02 a0.03 ± 0.01 c0.00 ± 0.00 c0.04 ± 0.02 c Chitinophaga0.28 ± 0.18 a0.00 ± 0.00 b0.03 ± 0.00 b0.03 ± 0.01 b0.07 ± 0.04 ab 浮霉菌门小计4.64 ± 0.88 a4.55 ± 0.51 a3.86 ± 0.25 a3.96 ± 0.33 a3.09 ± 0.92 a 小梨形菌属0.85 ± 0.28 a0.39 ± 0.03 ab0.44 ± 0.03 ab0.64 ± 0.09 ab0.38 ± 0.16 ab 浮霉状菌属0.62 ± 0.15 a0.74 ± 0.08 a0.61 ± 0.03 a0.17 ± 0.01 b0.48 ± 0.13 a 如出芽菌属0.66 ± 0.16 a0.50 ± 0.04 a0.21 ± 0.03 b0.17 ± 0.04 b0.11 ± 0.05 b Pir4 lineage0.16 ± 0.05 bc0.33 ± 0.05 a0.16 ± 0.01 bc0.01 ±0 .01 d0.19 ± 0.06 b Singulisphaera0.13 ± 0.03 b0.08±0.02 b0.13 ± 0.01 b0.29 ± 0.04 a0.09 ± 0.04 b 蓝藻门小计1.95 ± 1.14 ab1.42 ± 0.29 b1.07 ± 0.21 b3.14 ± 0.83 ab7.07 ± 3.68 a 鞘丝藻属0.03±0.02 a0.00 ± 0.00 a0.04 ± 0.02a0.00 ± 0.00 a0.02 ± 0.00 a 微鞘藻属0.01 ± 0.00 a0.04 ± 0.04 a0.00 ± 0.00 a0.00 ± 0.00 a0.01 ± 0.00 a 念珠藻属0.00 ± 0.00 b0.03 ± 0.01 a0.00 ± 0.00 b0.00 ± 0.00 b0.00 ± 0.00 b 席藻属0.00 ± 0.00 a0.03 ± 0.02 a0.01 ± 0.01 a0.00 ± 0.00 a0.00 ± 0.00 a Chlorogloeopsis0.00 ± 0.00 b0.04 ± 0.02 a0.00 ± 0.00 b0.00±0.00 b0.00 ± 0.00 b Persicaria minor0.00 ± 0.00 b0.00 ± 0.00 b0.00 ± 0.00 b0.01 ± 0.00 a0.01 ± 0.01 a 疣微菌门小计0.74 ± 0.06 b1.37 ± 0.07 a1.24 ± 0.19 ab0.80 ± 0.04 b0.71 ± 0.13 b 丰佑菌属0.64 ± 0.02 b1.29 ± 0.07 a1.14 ± 0.21 ab0.74 ± 0.04 ab0.57 ± 0.10 b 交替球菌属0.00 ± 0.00 c0.06 ± 0.01 ab0.02 ± 0.01 bc0.02 ± 0.00 bc0.07 ± 0.03 a 紫红球菌属0.00 ± 0.00 a0.00 ± 0.00 a0.00 ± 0.00 a0.00 ± 0.00 a0.00 ± 0.00 a 疣微菌属0.00 ± 0.00 a0.00 ± 0.00 a0.00 ± 0.00 a0.00 ± 0.00 a0.00 ± 0.00 a 真菌子囊菌门小计60.35±7.15b52.39±0.73b66.79 ± 4.83b87.67 ± 1.11 a85.02 ± 5.33 a 青霉菌属0.83 ± 0.38 b6.80 ± 0.45 b18.04 ± 1.84 ab28.52 ± 4.92 a1.86 ± 0.27 b 曲霉属0.95 ± 0.25 c4.45 ± 0.65 bc2.36 ± 0.77 bc13.87 ± 2.83 a2.16 ± 0.87 bc Mycothermus0.00 ± 0.0 0 c6.13 ± 1.00 a0.11 ± 0.02 bc0.00 ± 0.00 c1.44 ± 0.44 b 支顶孢属0.59 ± 0.31 a0.30 ± 0.03 a0.70 ± 0.24 a1.47 ± 0.27 a1.18 ± 0.53 a 镰刀菌属0.81 ± 0.37 ab0.21 ± 0.07b1.85 ± 0.86 a1.10 ± 0.22 ab0.13 ± 0.09 b Westerdykella0.06 ± 0.01 b3.62 ± 0.90 a0.04 ± 0.02 b0.04 ± 0.02 b0.03 ± 0.02 b 接合菌门小计5.25 ± 1.51 ab7.79 ± 1.38 a7.41 ± 2.15 a2.64 ± 0.39 b1.88 ± 0.66 b 被孢霉属5.25 ± 1.51 abc7.26 ± 1.18 ab7.41 ± 2.15 a2.63 ± 0.38 cd0.58 ± 0.29 d 毛霉菌属0.00 ± 0.00 a0.00 ± 0.00 a0.00 ± 0.00 a0.00 ± 0.00 a0.00 ± 0.00 a 担子菌门小计18.23 ± 9.61a15.47 ± 5.73 a7.20 ± 2.34 a3.06 ± 0.77 a5.32 ± 1.72 a Phylloporus0.90 ± 0.29 a2.72 ± 2.48 a0.17 ± 0.09 a0.07 ± 0.05 a0.33 ± 0.17 a 隐球菌属0.11 ± 0.10 b0. ± ±0.26 b0.22 ± 0.04 b0.23 ± 0.11 b0.67 ± 0.44 b Pseudozyma0.85 ± 0.69 a0.19 ± 0.06 a0.63 ± 0.20 a0.51 ± 0.03 a0.26 ± 0.11 a 鬼伞属0.01 ± 0.01 a0.02 ± 0.01 a1.50 ± 1.49 a0.00 ± 0.00 a0.00 ± 0.00 a 马拉色氏霉菌属0.04 ± 0.02 b0.01 ± 0.00 b0.01 ± 0.00 b0.01 ± 0.00 b0.03 ± 0.03 b Bjerkandera0.08 ± 0.07 a0.05 ± 0.03 a0.07 ± 0.02 a0.30 ± 0.27 a0.02 ± 0.01 a 壶菌门小计2.10 ± 0.69 b6.16 ± 2.29 a0.30 ± 0.11 b0.04 ± 0.02 b1.07 ± 0.34 b Spizellomyces0.00 ± 0.00 b0.39 ± 0.16 a0.00 ± 0.00 b0.00 ± 0.00 b0.00 ± 0.00 b Pateramyces0.01 ± 0.00 b0.24 ± 0.06 a0.07 ± 0.06 b0.00 ± 0.00 b0.00 ± 0.00 b Rhizophlyctis0.00 ± 0.00 a0.00 ± 0.00 a0.01 ± 0.01 a0.01 ± 0.01 a0.00 ± 0.00 a 根生壶菌属0.02 ± 0.02 a0.00 ± 0.00 a0.00 ± 0.00 a0.00 ± 0.00 a0.00 ± 0.00 a 球囊菌门小计0.08 ± 0.02 ab0.19 ± 0.10 a0.00 ± 0.00 b0.00 ± 0.00 b0.00 ± 0.00 b Paraglomus0.00 ± 0.00 b0.19 ± 0.10 a0.00 ± 0.00 b0.00 ± 0.00 b0.00 ± 0.00 b 球囊霉属0.07 ± 0.01 a0.00 ± 0.00 b0.00 ± 0.00 b0.00 ± 0.00 b0.00 ± 0.00 b 其他小计13.99 ± 3.8018.00 ± 2.0518.30 ± 1.926.59 ± 0.466.71 ± 2.72
2.5 草莓根际土壤微生物类群分析
在97% 的相似度下,得到了每个样品的OTU个数,CK、BM、S、G、FJB土壤分别获得1 858、2 058、2 064、1 627和1 410个细菌OTU以及350、368、380、219和208个真菌OTU。Venn图能够反映组间或样品之间共有和特有OTU数目,能够直观地表现出组间或样品间OTU的重叠情况。从图2中可以看出,不同地区之间共有806个细菌OTU以及107个真菌OTU,CK、BM、S、G、FJB土壤各自独有202、274、147、386、190个细菌OTU以及206、172、141、76、71个真菌OTU。
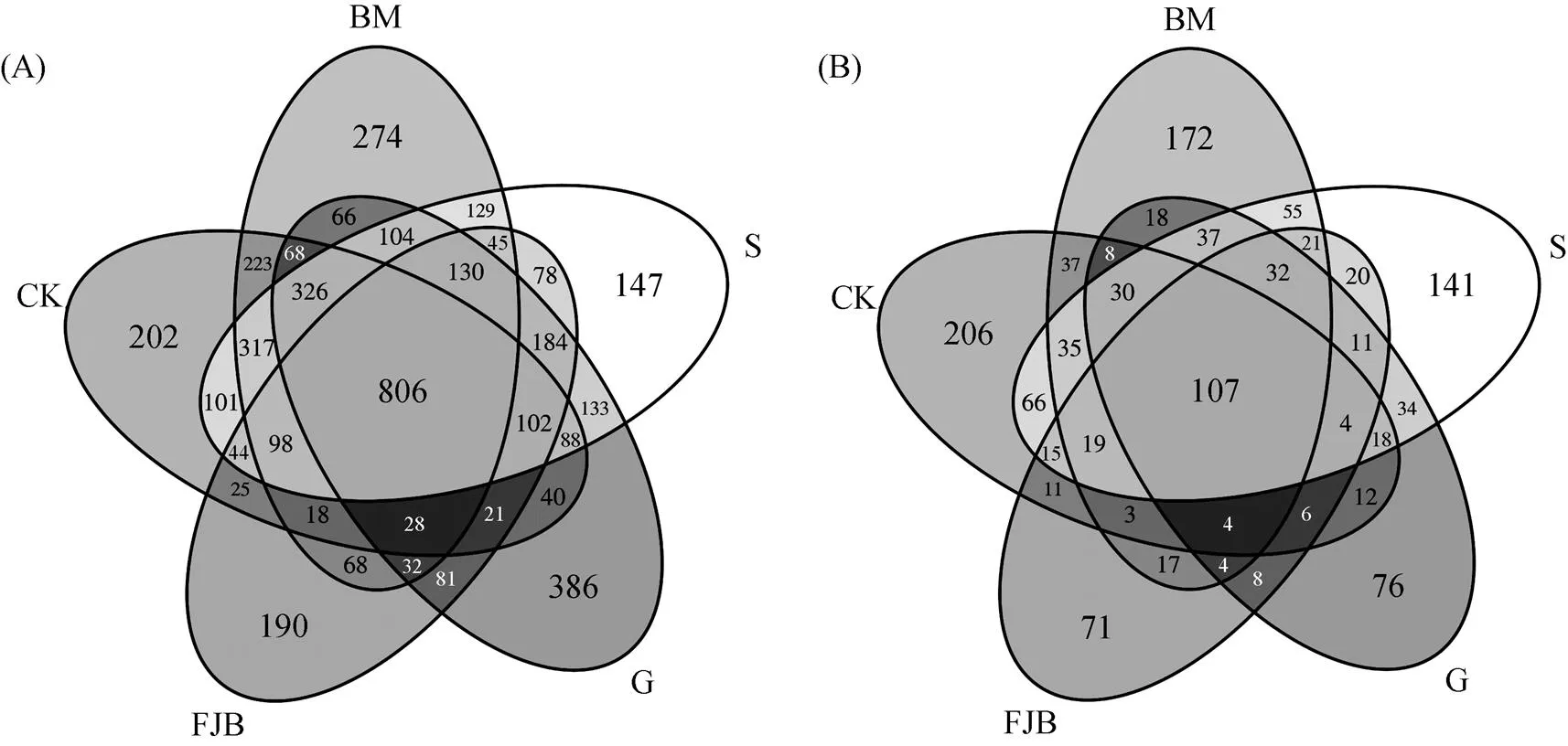
图2 草莓根际土壤细菌(A)、真菌(B) Venn图
2.6 土壤理化性质对微生物群落的影响
为了解土壤理化因子对土壤微生物群落的影响,在门水平上对土壤微生物群落进行冗余分析(RDA)(图3)。RDA分析的前两个轴总共解释了78.8% 的群落变化,第一个轴解释了55.0%,第二个轴解释了23.8%。土壤全氮对土壤微生物群落的影响最显著,解释了41% 的群落变化;其次是土壤pH,解释了20% 的群落变化,所有理化因子总共解释了90% 的群落变化,影响的顺序为土壤全氮>pH>有效磷>全钾>全磷>有机质>速效钾>碱解氮。
为了解环境因子与土壤微生物群落的关系,对土壤中的优势菌群(门水平)和土壤理化性质进行相关分析(表4)。结果表明:除变形菌门、放线菌门和酸杆菌门与所有理化指标相关性均不显著外,其他优势菌群都与土壤化学性质存在显著相关性。厚壁菌门与全氮、全磷、速效钾和有效磷含量呈显著负相关(<0.05),与有机质含量呈极显著负相关(<0.01);绿弯菌门与全氮含量呈显著正相关(<0.05),与全磷、碱解氮和有效磷含量呈极显著正相关(<0.01);子囊菌门与碱解氮含量呈显著负相关(<0.05),与全氮、全磷、速效钾、有效磷含量及pH呈极显著负相关(<0.01);担子菌门与全钾含量呈显著负相关(<0.05),与全氮、速效钾含量及pH呈极显著正相关(<0.01);除了全钾含量,接合菌门与其他土壤理化因子均呈显著正相关(<0.05)。
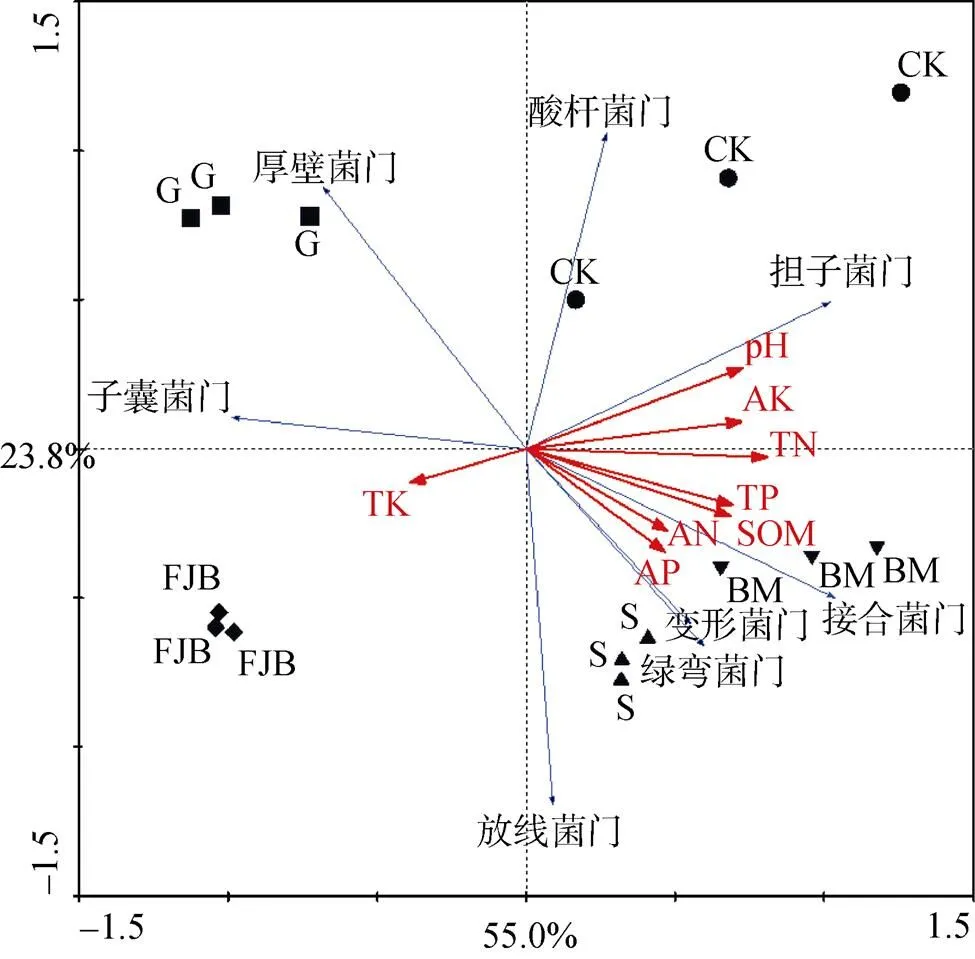
(图中AN、AP、AK、TN、TP、TK、SOM分别表示土壤碱解氮、有效磷、速效钾、全氮、全磷、全钾、有机质)
3 讨论
3.1 土壤微生物群落结构特征
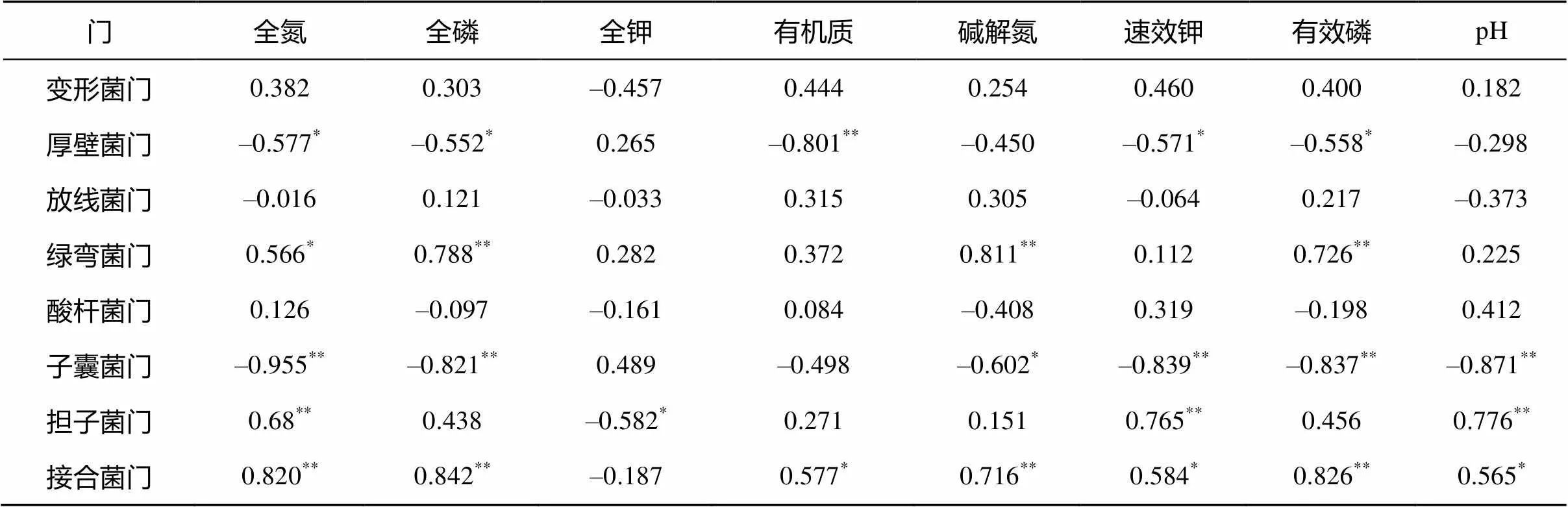
表4 土壤优势菌群(门水平)与土壤理化性质的相关性
注:*表示在<0.05水平上显著相关,**表示在<0.01 水平上显著相关。
土壤微生物群落是土壤生态功能的基础,通过参与土壤有机质分解和矿化等过程影响着土壤养分的循环并调节和指示土壤功能[14]。土壤中存在大量的微生物,其中细菌的种类和数量最多[15],细菌在有机物质的形成和分解、土壤营养元素的循环、肥力的保持和提高、植物的生长发育及病虫害防治、生态环境的改善等方面均起着极其重要的作用[16]。测序结果表明变形菌门、厚壁菌门、放线菌门、酸杆菌门和绿弯菌门为草莓根际土壤细菌的优势菌种,其中变形菌门的相对丰度最高,为23.51% ~ 35.46%。目前已在拟南芥[17]、玉米[18]、枸杞[19]根际中发现变形细菌的富集分布,可见变形细菌能够适应多种植物根际微环境。酸杆菌门作为优势菌种的相对丰度为6.4% ~ 15.14%,与柳春林等[20]和Peralta等[21]研究结果一致。酸杆菌门能够降解复杂的木质素[22]和纤维素[23]从而为土壤提供养分,残留的草莓植株经过酸杆菌门的分解为土壤微生物提供了充足的能源[24]。通过对优势菌种的分析,溧水白马、邳州港上、东海石榴、溧水傅家边4个区域的酸杆菌门的相对丰度都低于对照,这说明草莓连作会影响酸杆菌门细菌的生长发育,从而影响土壤养分。
土壤真菌是土壤微生物的重要成员,并与其他微生物一起推动着整个陆地生态系统的能量流动和物质循环,维持着生态系统的正常运转[25],是生态系统中极其重要的组成部分。测序结果表明,从草莓根际土壤中鉴定得到的真菌主要来自5个门,包括子囊菌门、接合菌门、担子菌门、壶菌门和球囊菌门。其中,子囊菌门、接合菌门和担子菌门为草莓根际土壤的优势菌种。子囊菌和担子菌是土壤中重要的分解者[26],子囊菌门大多数为腐生菌,可以分解很多难降解的有机质,如木质素和角质素,在养分循环中担任着重要角色[27]。测序结果还表明,镰刀菌属的相对丰度为0.13% ~ 1.85%,而镰刀菌属能够侵染包括草莓的多种植物(粮食作物、经济作物、油料作物、药用植物及观赏植物),致使植物发生萎焉、穗腐、根腐等各种类型的腐烂病,导致严重的减产,造成重大的经济损失[28]。
3.2 土壤微生物群落结构与环境因子的关系
土壤微生物群落结构和多样性受多种因素的影响,外因包括气候条件、植被类型、土壤类型和人类活动等,另一些是内因,主要是与土壤微生物生长密切相关的土壤有机质组成以及土壤养分等[29]。土壤微生物群落多样性分析(表2)表明,土壤细菌群落物种多样性的变化在不同区域间以及与对照间差异都不显著,而真菌群落物种多样性在区域间以及与对照间存在差异,这说明土壤真菌群落多样性比细菌群落多样性更受植物及其耕作方式等的影响。土壤微生物群落与土壤理化因子的冗余分析(图3)表明,土壤全氮对微生物群落的影响最显著,这与Liu等[30]的研究结果是一致的。氮肥影响了土壤微生物群落的结构组成,随着氮肥浓度的增加,富养性的分类群落变形菌门丰度增加[31]。其次,pH对微生物群落的影响最显著。土壤pH是影响微生物群落结构组成最重要的因素之一[32]。土壤其他理化因子对微生物群落结构都有重要的影响,说明土壤微生物群落结构与土壤环境关系密切。
植物种类对土壤微生物群落结构也有重要影响,相关研究表明植株根系分泌物对根际微生物群落结构组成具有选择塑造作用,不同植物的根际微生物群落结构具有独特性和代表性[33]。草莓的根系分泌物中含有乳酸、琥珀酸、己二酸、苯甲酸和对羟基苯甲酸,其中苯甲酸对根重具有明显的抑制作用,对草莓生长抑制作用最强[34]。化感物质可以单独对草莓植株造成危害,也可与其他病原复合作用于草莓,同时能够促进连作土壤中病原菌菌丝生长、孢子萌发及侵染,加重连作草莓根部病害,最终导致了草莓连作病害的发生[35]。近年来越来越多的学者认为,根系分泌物生态效应的间接作用以及土壤微生物区系紊乱是导致植物连作障碍发生的主要因素[36]。这可能是由于在根系分泌物特定组分的介导下,某些类群的微生物(如土传病原菌)大量繁殖,同时抑制其他有益微生物(如假单胞菌等拮抗菌)的生长,进而改变了植物根系分泌物的组分和数量,为趋化性病原微生物提供更多的碳源和能源,形成恶性循环,最终造成植物生长发育不良[37]。
4 结论
1) 草莓根际土壤的优势细菌门为变形菌门、厚壁菌门、放线菌门、酸杆菌门和绿弯菌门,主要的优势细菌属有16种;优势真菌门为子囊菌门、接合菌门和担子菌门,主要的优势真菌属有8种。
2) 微生物群落结构受多种土壤环境因子的影响,本研究中土壤全氮和pH对微生物群落的影响最显著。
[1] 于立杰, 梁春莉, 于强波. 草莓连作障碍发生机理及防治措施[J].安徽农业科学, 2009, 37(27): 13118–13119
[2] 汪其同, 朱婉芮, 刘梦玲, 等. 基于高通量测序的杨树人工林根际和非根际细菌群落结构比较[J]. 应用与环境生物学报, 2015, 21(5): 967–973
[3] Aira M, Bybee S, Domínguez J. Carnivory does not change the rhizosphere bacterial community of the plant Drosera intermedia[J]. Applied Soil Ecology, 2015(92): 14–17
[4] Acosta-martinez V, Burow G, Zobeck T M, et al. Soil microbial communities and function in alternative systems to continuous cotton[J]. Soil Science Society of America Journal[J], 2010, 74(4): 1181–1192
[5] Nayyar A, Hamel C, Lafond G, et al. Soil microbial quality associated with yield reduction in continuous-pea[J]. Appl Soil Ecology, 2009, 43(1): 115
[6] Schirmer M, Ijaz U Z, D'Amore R, et al. Insight into biases and sequencing errors for amplicon sequencing with the Illumina MiSeq platform[J]. Nucleic Acids Research, 2015, 43(6): e37
[7] Williams S T, Foster P G, Littlewood D T J. The complete mitochondrial genome of a turbinid vetigastropod from MiSeq Illumina sequencing of genomic DNA and steps towards a resolved gastropod phylogeny[J]. Gene, 2014, 533(1): 38–47
[8] Sogin M L, Morrison H G, Huber J A, et al. Microbial diversity in the deep sea and the underexplored “rare biosphere”[J]. Proceeding of the National Academy of Sciences of the United States of America, 2006, 103(32): 12115–12120
[9] Konstantinidis K T, Tiedje J M. Genomic insights that advance the species definition for prokaryotes[J]. Proceeding of the National Academy of Sciences of the United States of America, 2005, 102(7): 2567–2572
[10] Elad Y, Chet I, Henis Y. Biological control ofin strawberry fields by[J]. Plant and Soil, 1981, 60(2): 245–254
[11] 杨剑虹. 土壤农化分析与环境监测[M]. 北京: 中国大地出版社, 2008. pp 26–75
[12] Kozich J J, Westcott S L, Baxter N T, et al. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform[J]. Applied and Environmental Microbiology, 2013, 79(17): 5112–5120
[13] Amato K R, Yeoman C J, Kent A, et al. Habitat degradation impacts black howler monkey () gastrointestinal microbiomes[J]. The ISME Journal, 2013, 7(7): 1344–1353
[14] Romaniuk R, Giuffré L, Costantini A, et al. Assessment of soil microbial diversity measurements as indicators of soil functioning in organic and conventional horticulture systems[J]. Ecological Indicators, 2011, 11(5): 1345–1353
[15] 陈秀蓉, 南志标. 细菌多样性及其在农业生态系统中的作用[J]. 草业科学, 2002, 19(9): 34–38
[16] Kennedy A C. Bacterial diversity in agroecosystems[J]. Agriculture, Ecosystems and Environment, 1999, 74: 65–76
[17] Fierer N, Bradford M A, Jackson R B. Toward an ecological classification of soil bacteria[J]. Ecology, 2007, 88(6): 1354–1364
[18] Peiffer J A, Spor A, Koren O, et al. Diversity and heritability of the maize rhizosphere microbiome under field conditions[J]. Proceedings of the National Academy of Sciences, 2013, 110(16): 6548–6553
[19] 纳小凡, 郑国琦, 彭励, 等. 不同种植年限宁夏枸杞根际微生物多样性变化[J]. 土壤学报, 2016, 53(1): 241–252
[20] 柳春林, 左伟英, 赵增阳, 等. 鼎湖山不同演替阶段森林土壤细菌多样性[J]. 微生物学报, 2012, 52(12): 1489– 1496
[21] Peralta R M, Ahn C, Gillevet P M. Characterization of soil bacterial community structure and physicochemical properties in created and natural wetlands[J]. Science of the Total Environment, 2013, 443(3): 725–732
[22] Lynd L R, Weimer P J, VanZyl W H, et al. Microbial cellulose utilization: Fundamentals and biotechnology[J]. Microbiology and Molecular Biology Reviews, 2002, 66(3): 506–577
[23] Pankratov T A, Ivanova A O, Dedysh S N, et al. Bacterial populations and environmental factors controlling cellulose degradation in an acidic Sphagnum peat[J]. Environmental Microbiology, 2011, 13(7): 1800–1814
[24] Lopez-Mondejar R, Voriskova J, Vetrovsky T, et al. The bacterial community inhabiting temperate deciduous forests is vertically stratified and undergoes seasonal dynamics[J]. Soil Biology and Biochemistry, 2015, 87: 43–50
[25] 赵吉, 廖仰南. 羊草草地分解者亚系统的特性及作用[J]. 生态学报, 1995, 15(4): 359–364
[26] Yelle D J, Ralph J, Lu F C, et al. Evidence for cleavage of lignin by a brown rot basidiomycete[J]. Environmental Microbiology, 2008, 10(7): 1844–1849
[27] Beimforde C, Feldberg K, Nylinder S, et al. Estimating the phanerozoic history of the ascomycota lineages: Combining fossil and molecular data[J]. Molecular Phylogenetics and Evolution, 2014, 78: 386–398
[28] 张向民. 镰刀菌属分类学研究历史与现状[J]. 菌物研究, 2005, 3(2): 59–62
[29] 杨瑞红, 赵成义, 王新军, 等. 梭梭和柽柳土壤微生物多样性初步分析[J]. 土壤, 2016, 48(6): 1120–1130
[30] Liu J, Sui Y, Yu Z, et al. High throughput sequencing analysis of biogeographical distribution of bacterial communities in the black soils of northeast China[J]. Soil Biology & Biochemistry, 2014, 70(2): 113–122
[31] Fierer N, Lauber C L, Ramirez K S, et al. Comparative metagenomic, phylogenetic and physiological analyses of soil microbial communities across nitrogen gradients[J]. The Isme Journal, 2012, 6(5): 1007
[32] 纳小凡, 郑国旗, 邢正操, 等. 连作对再植枸杞根际细菌群落多样性和群落结构的影响[J]. 土壤学报, 2017, 54(5): 1280–1292
[33] Paterson E, Gebbing T, Abel C, et al. Rhizodeposition shapes rhizosphere microbial community structure in organic soil[J]. New Phytologist, 173, 600–610
[34] Kitazawa H, Asao T, Ban T, et al. Autotoxicity of root exudates from strawberry in hydroponic culture[J]. The Journal of Horticultural Science and Biotechnology, 2005, 80(6): 677–680
[35] 侯慧, 董坤, 杨智仙, 等. 连作障碍发生机理研究进展[J]. 土壤, 2016, 48(6): 1068–1076
[36] Qi J J, Yao H Y, Ma X J, et al. Soil microbial community composition and diversity in the rhizosphere of a Chinese medicinal plant[J]. Communications in Soil Science and Plant Analysis, 2009, 40(9/10): 1462–1482
[37] 吴林坤, 林向民, 林文雄. 根系分泌物介导下植物-土壤-微生物互作关系研究进展与展望[J]. 植物生态学报, 2014, 38(3): 298–310
Microbial Community Structures and Diversities in Strawberry Rhizosphere Soils Based on High-throughput Sequencing
ZHAO Fan1,2, ZHAO Mizhen1*, WANG Yu2, GUAN Ling1, PANG Fuhua1
(1 Institute of Pomology, Jiangsu Academy of Agricultural Sciences & Jiangsu Key Laboratory for Horticultural Crop Genetic Improvement, Nanjing 210014, China; 2 College of Resources and Environment Engineering, Anhui University, Hefei 230601, China)
It is important for constructing and maintaining a healthy soil ecosystem to explore microbial community composition and structure in strawberry rhizosphere soil. Strawberry rhizosphere soils in different places were collected and used as study targets, the 16S rRNA genes of V4+V5 regions of soil bacteria and of ITS1+ITS2 regions of soil fungi were sequenced and analyzed by Illumina high-throughput sequencing technology on Miseq platform combined with related bioinformatics analysis to explore the changes of abundances, diversities and structures of soil bacteria and fungi. Results showed that a total of 4 554 bacterial operational taxonomic units (OTUs) and 1 298 fungal OTUs were obtained from 15 strawberry rhizosphere soil samples. At phylum level, dominant bacteria were Proteobacteria, Firmicutes, Actinobacteria, Acidobacteria and Chloroflexi, dominant fungi were Ascomycota, Zygomycota and Basidiomycota. At genera level, there were 16 genera of dominant bacteria and 8 genera of dominant fungi. Redundancy analysis (RDA) showed that total nitrogen and pH had the greatest effect on soil microbial community structure, explaining 61% of the community changes. The order of contribution rate was total nitrogen > pH > available phosphorus > total potassium > total phosphorus > organic matter > available potassium > alkali-hydrolyzable nitrogen. Correlation analysis also showed that soil physiochemical characteristics were significantly correlated with different dominant microbial community. This study deepens the understanding on microbial community in strawberry rhizosphere and provides references for the relation between microbial composition and diversity with environmental factors.
Strawberry; Rhizosphere soil; Illumina high-throughput sequencing; Soil microbial community; Soil physiochemical characteristics
江苏省现代农业研究开发示范类项目(BE2016369)和江苏省农业三新工程项目(SXGC[2017]261)资助。
通讯作者(njzhaomz@163.com)
赵帆(1993—),男,江苏南京人,硕士研究生,主要从事农业生态系统研究。E-mail: 843003818@qq.com
S663.9
A
10.13758/j.cnki.tr.2019.01.008
猜你喜欢
当代水产(2022年8期)2022-09-20
浙江农业科学(2022年7期)2022-07-15
中国农学通报(2022年14期)2022-06-01
油气田环境保护(2022年2期)2022-05-09
昆明医科大学学报(2022年2期)2022-03-29
中南林业科技大学学报(2021年11期)2021-12-17
中国土壤与肥料(2021年5期)2021-12-02
四川农业科技(2021年7期)2021-12-01
食品安全导刊(2021年20期)2021-08-30
中国果业信息(2020年10期)2020-12-15
