
11例儿童RAS通路病的临床特点和基因突变类型
2019-04-08 09:04:04江转南刘祖霖张丽娜侯乐乐黄思琪宋青芳梁立阳
中山大学学报(医学科学版) 2019年2期
江转南,刘祖霖,张丽娜,侯乐乐,孟 哲,黄思琪,宋青芳,梁立阳
(中山大学孙逸仙纪念医院儿科,广东广州510120)
RAS通路病是一组以丝裂原活化蛋白激酶通路异常(RAS/mitogen-activated protein kinase,RAS/MAPK)为共同发病机制的遗传性疾病。在RAS/MAPK通路中编码激活酪氨酸激酶受体(receptor tyrosine kinases,TRK)、RAS蛋白、RAS功能的调节器以及下游信号传感器的基因出现突变均会导致RAS通路病,包括7种亚型:1型神经纤维瘤病(neurofibromatosis type 1,NF1)、努南综合征(Noonan syndrome,NS)、多色素痣型努南综合征(Noonan syndrome with multiple lentigines,NSML)、毛细血管-动静脉畸形综合征(capillary malformation-arteriovenous malformation syndrome,CMAM),科斯特洛综合征(Costello syndrome,CS),心脏-皮肤-面综合征(cardio-facio-cutaneous syndrome,CFC)和类1型神经纤维瘤病(Legius syndrome,LS)[1]。RAS通路病总发病率为1/1 000,其中以NS最常见,发病率达1/1 000~1/2 500,其次是1型神经纤维瘤病,发病率约1/3 000,但CS和CFC非常罕见,发病率分别为1∶1 290 000和1∶810 000,NSML尚无发病率统计[2]。现将我科确诊的11例RAS通路病患者的临床资料和基因变异类型结合国内外相关文献进行总结和分析。
1 材料与方法
1.1 临床资料收集
收集2015年10月至2018年6月来我院就诊并经基因测序确诊的11例RAS通路病患儿的临床资料,包括家族遗传史、出生史、个人生长发育史、既往疾病史、实验室检测结果、基因检测结果、影像学结果等。本研究经医院医学伦理委员会批准,所有受试患儿家属均知情同意并签署知情同意书。
1.2 实验方法
1.2.1 实验室检测 所有患儿均进行血、尿、粪便常规检查、电解质、肝肾功能、心肌酶谱等,部分患儿根据症状选择性行心脏超声、头颅核磁共振、脑电图和生长激素激发试验等辅助检查,了解病情并排除其他病因。
1.2.2 基因检测 分别抽取患儿及其父母静脉血各2 mL,送至广州金域医学检验中心公司进行基因测序。
1.2.3 突变分析 对于测序结果中的变异位点,参照人类孟德尔遗传数据库(OMIM)、寡核苷酸多态性数据库(dbSNP)、人类基因突变数据库(HGMD)、ClinVar数据库和DrugBank数据库等确定变异位点性质。
2 结果
2.1 患儿临床及实验室检查结果
本文11例RAS通路病患儿包括3例NF1、4例NS、1例NSML、2例CS和1例CFC,未包括CMAM和LS。初诊年龄最小者6个月,最大者12岁,男性6例,女性5例。主要症状包括身材矮小、特殊面容、先天性心脏病、牛奶咖啡斑、运动发育落后、血小板减少、抽搐、肌张力异常、隐睾等。特殊面容特征包括卷发、前额突出、眼距宽、鼻梁塌陷和低耳位等(图1)。先天性心脏病包括动脉导管未闭、房间隔缺损、肺动脉狭窄和卵圆孔未闭。11例患儿的临床资料和实验室检查结果见表1。
2.2 治疗与随访
3例NS患儿正在进行重组人生长因子(recombinant human growth hormone,rh-GH)治疗,目前疗效理想,暂无不良反应,除1例NS患者有手部良性肿物切除史暂无复发外,余3例在随访中尚未发现肿瘤生长。2例CS患儿目前在随访过程中均未发现肿瘤生长,针对语言、运动发育迟缓进行康复训练、口服神经营养药物干预治疗和个性化教育,患儿的运动及智力均进步,但身高生长速度明显落后。1例NF1患儿口服奥卡西平抗癫痫治疗,另1例NF1患儿口服苯海索调节肌张力,疗效均可。余1例NF1、1例CFC以及1例NSML患儿目前仍在随访监测中,暂未发现肿瘤生长。
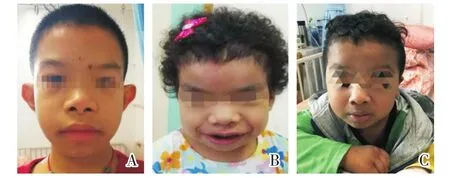
图1 3例RAS通路病患儿的面部特征Fig.1 Craniofacial features of three RASopathies patients
2.3 基因突变类型与家系分析
基因检测结果显示(表2):突变基因类型包括NF1基因、PTPN11基因、RAF1基因、BRAF基因和HRAS基因;不同的突变位点共9个;8例为新发突变,3例遗传自父母。其中NS.1与NS.2患儿是兄妹关系,NS.1示先证者,其父亲有严重矮小表型,3者携带相同的突变位点,追溯其父亲血缘成员均无异常表型。另外,NF.1患儿的突变位点遗传于母亲,其母亲为独生子女,有“神经纤维瘤”表现,但其外祖父母在其母亲2岁时已逝世(未透露逝世原因)。3例阳性家族遗传史患儿的家系家谱图如图2。
3 讨论
3.1 RAS通路病的临床共性
RAS通路病的共同发病机制为RAS/MAPK通路异常。RAS/MAPK通路在调控生长发育、促进细胞的增殖、分化、代谢以及多种激素细胞内外信号转导中发挥重要作用。因此,RAS通路病具有多种共同临床表现,包括特殊面容、身材矮小、运动发育落后、心血管、皮肤、神经等多系统异常以及肿瘤易感性等,临床实践中仅凭临床表现很难具体分型。但因基因突变位点的差异,各亚型间亦存在相对特异性表现[1-2]。
3.2 RAS通路病的临床特性
3.2.1 NS临床特性 NS是一种常染色体显性遗传病,1968年由Jacqueline Noonan首次报道[3]。主要特征包括身材矮小、运动发育落后、智力低下、特殊面容、多系统组织异常以及肿瘤易感性等[3-4]。NS患者的面部特征包括“三角形”脸型、前额宽、眼裂下斜、内眦赘皮、上睑下垂、耳位低、耳轮后旋、颈短和颈蹼等[2]。本文4例NS患儿均有特殊面容,尤以NS.4患儿的面部特征相对典型。NS患者多伴有口腔牙齿畸形,可表现为高腭弓、恒牙不长、外生牙畸形等[2]。80%以上的NS患者有心血管病变,以肺动脉狭窄最常见,其次是肥厚型心肌病[5]。本文3例NS患儿有先天性心脏病史,包括1例动脉导管未闭、1例房间隔缺损和1例房间隔缺损合并肺动脉狭窄术后。NS患者的皮肤病变多表现为皮肤色素沉着、卷发稀发、指甲营养不良等[6]。约80%男性NS患者伴有隐睾,部分存在生殖障碍,女性NS患者的生殖功能多数正常[7]。相对其他RAS通路病而言,血液系统异常是NS较为独特的临床表现,如血小板减少、凝血功能异常和骨髓增生异常等,可以为首发症状[8],本文2例NS患儿亦曾以血小板减少为主要症状就诊。另外,NS患者亦可有消化、神经和骨骼系统病变,表现为早期喂养困难、脾大、早期运动发育落后、癫痫、肌张力减低、鸡胸、漏斗胸、脊柱侧弯等[7,9-10]。目前已报道12种基因突变与NS以及NS样疾病的发病有关,较为常见的3种基因分别为PTPN11(>50%)、SOS1(约10%)和RAF1(5%~10%),另外 9种基因包括KRAS、NRAS、BRAF、SHOC2、CBL、MAP2K1、A2ML1、SOS2和RIT1等[1-2,11-12]。NS患者的基因型和临床表型存在一定的相关性,比如PTPN11基因突变者常伴有严重身材矮小和低胰岛素样生长因子-1水平;RAF1或RIT1基因突变者心血管异常发病率明显升高等[1,6-7]。目前,NS仍以临床诊断为主,主要参考1994年荷兰作者提出的诊断标准[13],而对于临床诊断有困难者,则可行基因检测以明确诊断。有关NS患者的治疗,以对症与随访为主,心血管或骨骼异常者需定期随访,必要时介入或外科手术治疗;国内外已推荐NS患者使用rh-GH促进生长发育。本文3例正在进行rh-GH治疗的NS患儿,尚无不良反应,疗效理想。
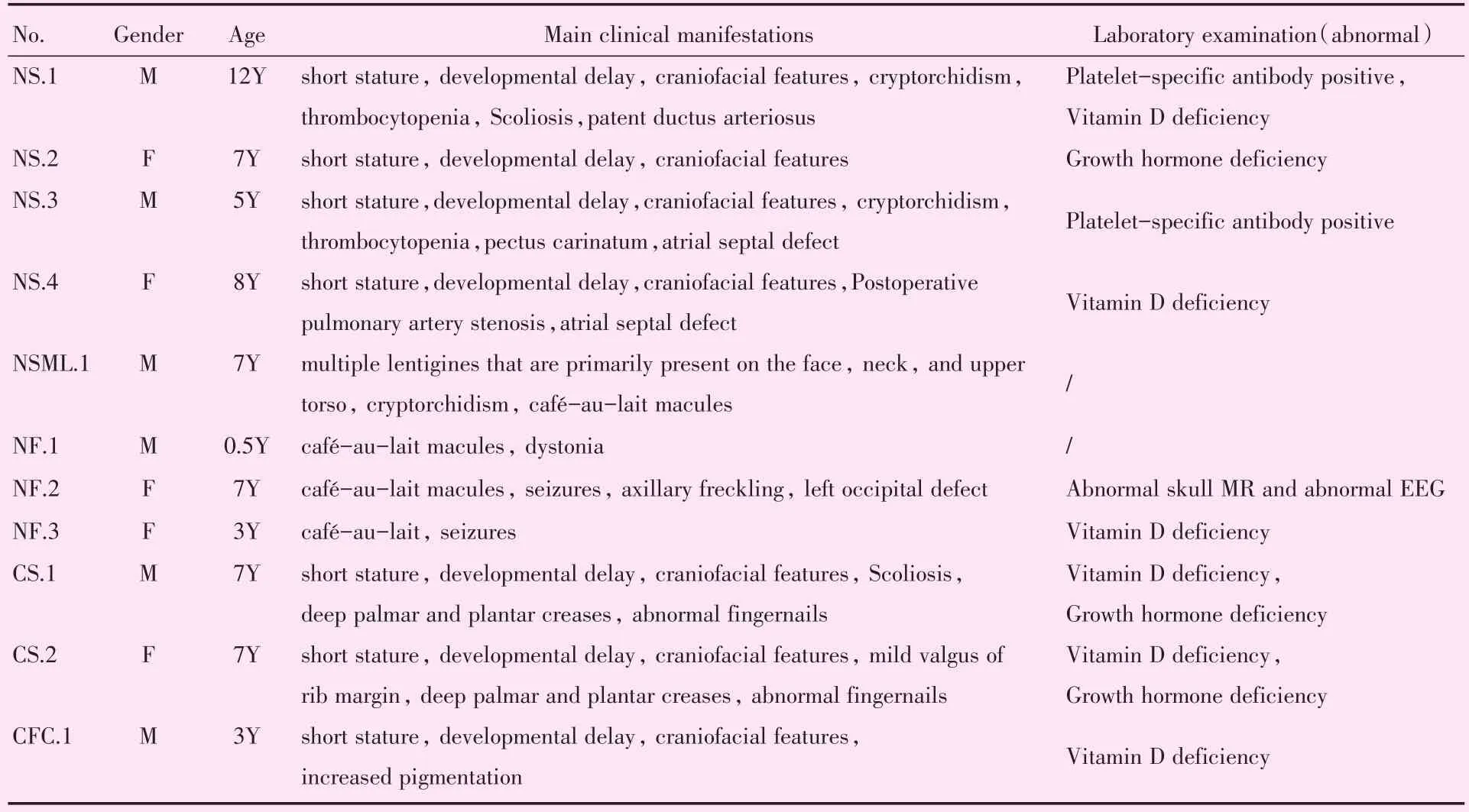
表1 患儿主要临床表现及实验室检查结果Table 1 The main clinical manifestations and laboratory examination of patients(n=11)
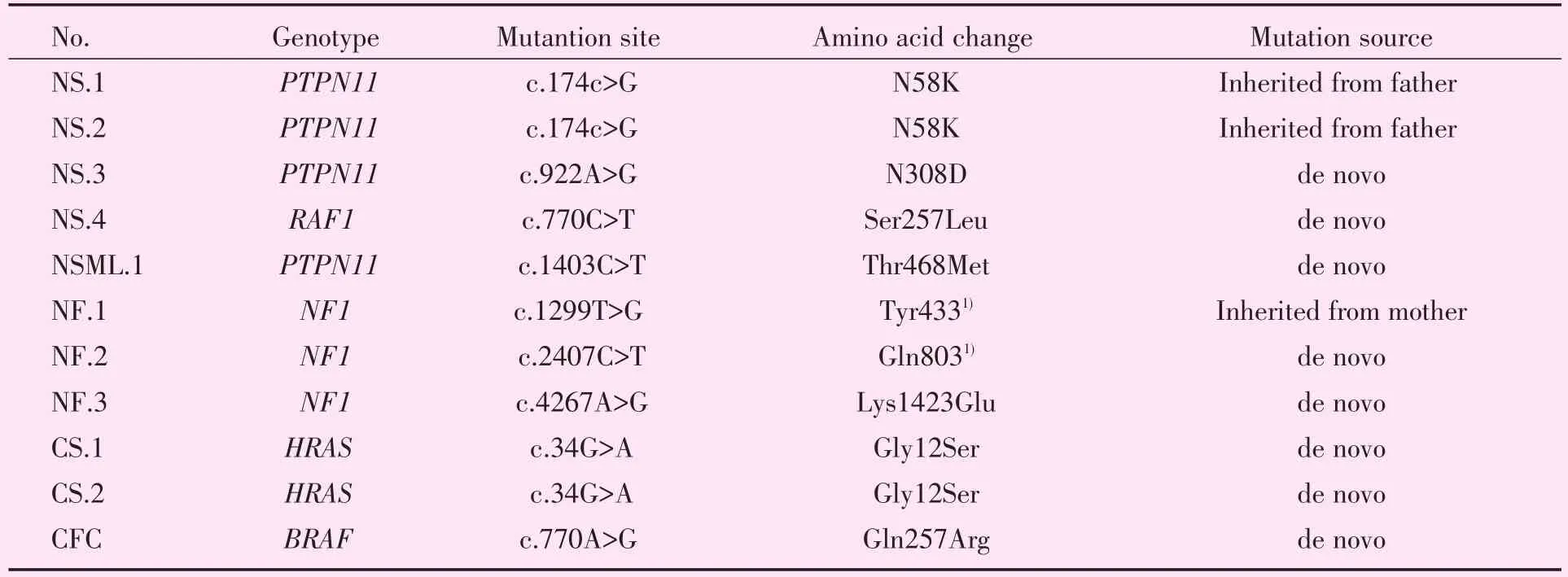
表2 RAS通路病患儿的基因突变结果Table 2 Gene mutation results of 11 cases of RASopathies patients

图2 3例阳性家族遗传史患儿的家系家谱图Fig.2 Family tree of 3 patient with positive family history
3.2.2 NSML临床特性 NSML是一种极罕见的常染色体显性遗传病,目前已报道的NSML病人数约200例[2]。NSML的面部特征与NS相似,但相对不典型,如下颌前凸、巨舌、不伴颈蹼等。NSML的典型特征则是颈部、面部、上躯干多发性色素痣,常于4~5岁时出现,儿童期呈指数增长,青春期后可达数千[2]。本文确诊的1例NSML的临床表现非常典型,全身色素痣随年龄增长,伴牛奶咖啡斑和隐睾。感音神经性耳聋在NSML患者的发病率远高于NS患者,发病率分别为NSML>20%和NS<5%[14]。在多种RAS通路病中,肥厚型心肌病的发病率在NSML患者中是最高的,达80%以上[15]。NSML的致病基因有3种,包括PTPN11基因、RAF1基因和BRAF基因[1-2],其中PTPN11基因和BRAF基因亦是NS的致病基因,因此两种综合征临床表现相似,但因突变位点不一样,两者亦有差异性。本文NSML患者的突变基因位点:PTPN11Exon12:c.1403C>T;p.(Thr468Met),该位点已有报道。
3.2.3 NF1临床特性 NF1是一种常染色体显性遗传病,典型临床特征包括多发神经纤维瘤、皮肤牛奶咖啡斑、虹膜色素错构瘤、视神经胶质瘤和骨骼发育不良等[1-2]。神经纤维瘤是NF1的特异性临床表现,属于周围神经鞘的良性肿瘤,主要发生在头颈区。NF1的临床诊断可参照NIH诊断标准[16],但对于不满足NIH诊断标准,但又高度怀疑为NF1的患者,基因检测可为诊断提供重要依据。NF1基因突变是NF1的重要发病机制,该基因编码的神经纤维瘤素是一种肿瘤抑制物,是RAS信号转导的一种负性调节因子。NF1基因突变导致神经纤维瘤素的负性调节作用消失,从而引起RAS的活性增高,最终导致异常的细胞增生和肿瘤生长[17]。目前,NF1以手术治疗为主,手术切除不会增加新的神经纤维瘤的出现几率,或者引起未完整切除部分发生恶变。神经纤维瘤的复发与年龄、病变部位和手术范围相关[18-19]。本文3例NF1患者均有典型皮肤牛奶咖啡斑表现;其中症状性癫痫表现者1例,头颅MR见多发异常信号影及左枕颞部分骨质缺如,脑电图清醒期见双侧额区、左侧颞区阵发同步性中高波幅尖波;3例身高正常或偏矮。
3.2.4 CS临床特性 CS于40余年前首次由儿科医生Costello JM报道,亦呈常染色体显性遗传。CS患者的面容相对粗陋,典型面部特征包括高前额、稀发卷发、内眦赘肉、鼻梁塌、厚唇大嘴,且多伴有掌跖部皮纹深大、皮肤松弛、色素沉着等皮肤异常特征[20-21]。胎儿期超声检查即可发现CS患者颈部厚度增加、手腕尺侧偏、短肱骨及羊水明显增多等影像学表征[22]。高达80%的患者伴有心血管病变,与NS患者的相似。CS患者出现癫痫发作可能性相对高,达20%~25%。关节松弛、先天性髋关节发育不良、手腕尺侧偏斜和跟腱紧绷等是CS患者较为特征性的骨骼病变[20],实验室检查亦可发现骨密度低和维生素D缺乏等。横纹肌肉瘤、神经母细胞瘤以及膀胱癌是CS患者常见的恶性肿瘤疾病,应注意定期监测。HRAS基因突变是CS的重要发病机制,目前已报道的突变位点已达10余种[23],本文2例CS患儿的突变位点一致,均为HRASxon2 :c.34G>A,p.Gly12Ser,该位点是最常见的突变类型,占80%以上,该突变可使RAS蛋白持续性激活,神经胶质细胞过度增殖,影响皮层的正常发育。CS的临床诊断相对困难,不易与其他RAS通路病鉴别,基因测序有助于明确诊断。定期随访及改善患者整体健康和智力发育是Costello综合征的治疗目的。
3.2.5 CFC临床特性 CFC亦是一种常染色体显性遗传病,首次由Reynolds于1986年报道[24],CFC患者的临床表现与NS、CS患者非常近似。CFC的面部特征包括相对大头畸形、额头高大、双颞侧变窄、眶上脊发育不良、鼻子基底部过宽、球状鼻头、人中沟深、耳位低、耳轮向后旋等[2];CFC患者常合并皮肤病变,如色素痣、皮肤角化过度症等,亦可出现斜视、眼球震颤等眼科症;而CFC患者的心血管病变亦与CS或NS相似。CFC患者常伴有神经系统、骨骼系统的病变。目前发现CFC患者有4种基因突变类型,其中BRAF基因突变占75%,MAP2K1/MAP2K2基因突变占25%,KRAS基因突变占2%~3%[25]。CFC亦有明显基因-表型相关性,BRAF突变患者可能更易出现心血管病变,如肺动脉狭窄、肥厚型心肌病以及房间隔缺损,可伴有中-重度智力障碍以及喂养困难;而MAP2K1/MAP2K2基因突变可能与早产、室间隔缺损、泌尿生殖系统或皮肤组织异常相关。因不易与NS、CS鉴别,基因测序有助于确诊CFC。CFC治疗原则与CS相似,以对症与监测为主。本文1例CFC患者有典型的临床表现,包括严重身材矮小、卷发、耳位低、运动语言落后以及皮肤黏膜色素沉着等,其基因突变位点BRAFExon6 c.770A>G p.(Gln257Arg),为新发突变,既往已有文献报道该致病位点。
上述5种亚型RAS通路病临床表现多样,同时具有多种临床共性和特性,其中NF1的临床表现相对特异,容易确诊,而另外4种综合征则可通过面容特征、皮肤异常表现、骨骼病变、先心病类型以及是否伴有隐睾或血液系统异常等差异进行初步临床鉴别,比如:80%男性NS患儿伴有隐睾表现;血液系统异常在NS患儿较为常见,如血小板减少、凝血功能异常和骨髓增生异常等,可以为首发症状;感音神经性耳聋和肥厚型心肌病在NSML患儿当中发生率高,且颈部、面部、上躯干可见多发色素痣;CS患儿面容丑陋,多伴有掌跖部皮纹深大、皮肤松弛、色素沉着等特异性皮肤表现以及关节松弛、手腕尺侧偏斜和跟腱紧绷等特异性骨骼病变;而新生儿难治性低血糖、喂养困难等则是CS和CFC患儿常见的早期表现;若诊断仍有困难,建议借助基因测序以明确诊断。因该类疾病在早期无明显特异性表现,加之临床医师对RAS通路病认识的不足,对于以新生儿低血糖、喂养困难为早期表现的婴幼儿,常常不能引起临床医生的警惕。多数患儿在学龄期前期或学龄期因身材矮小、智力低下或运动发育落后等就诊才得以确诊的。本文11例患儿中有7例患儿均以身材矮小、运动发育落后为主诉,初诊年龄中位数为7岁。学者们提倡该类疾病应早期确诊,对于临床上遇到的以顽固性低血糖、喂养困难、身材矮小、特殊面容、智力低下以及多系统异常为表现的患儿,须警惕RAS通路病可能,确诊后及时予对症支持治疗,包括早期联合营养科制定喂养计划改善患儿营养情况、婴幼儿期制定个性化运动及语言康复锻炼提高患儿生活水平等,此类患儿需定期随访,密切监测肿瘤、生长发育、心血管疾病等病情变化。
猜你喜欢
英语世界(2023年6期)2023-06-30 06:29:10
上海金属(2021年6期)2021-12-02 10:47:20
昆明医科大学学报(2021年3期)2021-07-22 07:40:04
中国生殖健康(2020年2期)2021-01-18 02:51:26
生物学通报(2019年3期)2019-02-17 18:03:58
小学生导刊(2018年13期)2018-06-29 03:49:00
中国病理生理杂志(2015年8期)2015-12-21 12:38:06
医学研究杂志(2015年3期)2015-06-10 06:41:52
创业家(2015年1期)2015-02-27 07:52:02
遗传(2014年2期)2014-02-28 20:58:11
