
高速逆流色谱结合半制备型液相色谱分离茶树紫芽花色苷研究
2019-04-06 11:56刘林峰肖文军龚志华
茶叶通讯 2019年2期
张 拓,刘林峰,林 玲,周 阳,肖文军,2,龚志华*
(1湖南农业大学 茶学教育部重点实验室,国家植物功能成分利用工程技术研究中心,湖南 长沙 410128;2湖南农业大学 湖南省植物功能成分利用协同创新中心,湖南 长沙 410128)
花色苷是一类具有2-苯基苯并吡喃基本母核结构的植物次生代谢产物,具有调节糖脂代谢、提高免疫、清除自由基、抗氧化等多种生物活性[1-4],是近年天然产物开发利用领域的热点之一。茶树芽叶出现紫化主要是由茶树种质、发育特性及其生长环境条件等因素所引起的花色苷含量显著增加的抗逆生理表现。在传统茶叶加工中,由于以紫色芽叶加工而成的茶产品在干茶色泽、汤色及叶底等品质因子上呈紫色,被认为不适于茶叶加工而造成大量浪费。随着紫芽茶树花色苷研究的深入以及健康意识的增强,茶叶花色苷的高值化利用越来越受到业界的关注。然而,由于茶叶原料中花色苷含量较低,且存在多种花色苷组分,从而使得制备高纯度的花色苷及其组分难度较大。目前分离纯化植物花色苷的技术方法主要有柱层析法、高速逆流色谱法、半制备型高效液相色谱法和反相高效液相色谱法等,如申芮萌等[5]采用葡聚糖Sephadex LH-20凝胶层析柱分离纯化蓝莓中的花色苷,并优化了分离花色苷的最佳缓冲液浓度、流速以及上样量等条件;曹少谦等[6]采用柱层析法分离纯化血橙中的花色苷,筛选出最佳的大孔树脂,同时对工艺进行优化;于泽源等[7]采用大孔树脂-中压柱层析分离纯化蓝莓花色苷,其纯度为90.88%;刘雪辉等[8]、陆英等[9]利用高速逆流色谱技术研究了玫瑰茄和紫甘薯中的花色苷分离纯化工艺,并对工艺进行优化;SAITO T[10]采用色谱法研究红芽茶树中花色苷的分离工艺;以及以半制备型高效液相色谱法和反相高效液相色谱技术来分离纯化花色苷[11],但大多研究局限单一技术方法分离纯化花色苷,结合不同分离技术的方法分离纯化茶叶花色苷及其组分的研究鲜见报道。本研究以茶树紫色芽叶为材料,经酸性乙醇避光浸提、萃取脱脂、阳离子交换树脂吸附等初步分离,探究了高速逆流色谱结合半制备型液相色谱制备高纯茶叶花色苷组分的工艺技术,以期为茶树紫色芽叶及其花色苷的开发利用提供参考。
1 材料和方法
1.1 茶叶与试剂
茶树鲜叶:采摘湖南农业大学长安实践教学基地高花色苷茶树品系自选9803的一芽二叶茶鲜叶,经冷冻干燥、粉碎后得茶叶粉末,低温密封保存备用。
药品试剂:乙腈、甲醇(色谱纯)、甲醇、无水乙醇(分析纯)、天津市恒兴化学试剂制造有限公司;正己烷、乙酸乙酯(分析纯)、国药集团化学试剂制造有限公司;矢车菊素-3-O-葡萄糖苷、天竺葵素-3-葡萄糖苷:北京谱析科技有限公司;飞燕草素-葡萄糖苷:西宝生物科技(上海)股份有限公司;超纯水:自制;酸性乙醇水溶液:自制;钠型732阳离子交换树脂:国药集团化学试剂制造有限公司。
1.2 仪器设备
超声波清洗器,KQ2200B型,昆山超声仪器有限公司;冷冻干燥机,MODULYOD-230型,Thermo Fisher Sciebti fic,美国;色谱分析柱(4.6 mm×250 mm,5 μm),C18 ECOSIL HPLC COLUMN型,日本ECOSIL公司;紫外可见分光光度计,UV754N型,德国Thermo Scienti fic公司;SPD-20A型紫外检测器;Unitary C18反相色谱柱 (10 mm×250 mm,5 μm);高效液相色谱仪,A20型,日本Shimazu公司;精密电子天平,AE240型,瑞士Starorius公司;pH计,瑞士梅特勒-托利多公司;超低温冰箱,美国Thermo公司;水浴锅,DSY-2-8型,北京国华医疗器械厂;高速逆流色谱(内径1.6 mm,容积 280 mL,转速 0~1000 r/min),TBE-300A 聚四氟乙烯柱,上海同田公司;制备型高效液相色谱仪,LC-8A 型,日本Shimadzu Corporation公司;旋转蒸发器,SY-2000型,上海荣亚生化仪器厂。
1.3 实验方法
1.3.1 茶树紫芽花色苷的提取
取茶样干粉21.96 g,花色苷提取条件参考文献[12-14],设置为:料液体积比1∶25、80%乙醇(加1%冰醋酸)、室温避光浸提24 h、提取两次,合并两次浸提液,蒸发浓缩至膏状后获得提取物保存备用。
1.3.2 脱脂
取茶样干粉13.60 g,配置正己烷∶乙酸乙酯∶水混合溶剂(1∶1∶2,V/V/V),对茶样提取液进行萃取脱脂处理,萃取至有机层无色为止,收集脱脂液,低温保存备用。
1.3.3 离子交换树脂分离
以1.3.2得到的脱脂液为上样液,经钠型732阳离子交换树脂[15-16]吸附,树脂前处理方法参照马淑青等[15],上样液pH 值为3、上样液流速为5 BV/h[17-18],吸附至饱和为止;吸附完成后分两次洗脱, 第一次洗脱:采用80%乙醇溶液洗脱,洗脱液流速3 BV/h,洗脱液合并收集10 BV浓缩至1 BV去有机相,得到浓缩液I,HPLC检测分析浓缩液I的浓度;第二次洗脱:依次采用2 BV不同比率4%盐酸-乙醇溶液洗脱,洗脱液流速3 BV/h,洗脱梯度为50%、60%、70%、80%、90%,按乙醇浓度分别收集洗脱液,2 BV合并浓缩至1 BV去有机相,得浓缩液Ⅱ,HPLC检测分析浓缩液Ⅱ的浓度。
1.3.4 大孔树脂除酸
离子交换树脂的第二次洗脱液浓缩后经D101大孔树脂吸附树脂去除盐酸,浓缩收集液、冷冻干燥得到紫芽茶树花色苷粗品粉末,粉末用甲醇溶解,HPLC检测并计算粗品总花色苷纯度(参照1.3.7)。
1.3.5 高速逆流色谱溶剂体系筛选与分离
按表1设计不同溶剂体系方案,按比例配置10 mL体系溶液。振荡混匀后待静置分层,移取3 mL下相加入少量上述花色苷粉末,再用3 mL上相萃取,分别检测萃取前后花色苷组分的峰面积,各组分的分配系数(K值)计算方法参考文献陈田等[19],筛选最佳溶剂体系。
选取表1中最佳溶剂体系方案,高速逆流色谱分离参数参考文献[8,19]:将最佳溶剂体系静置12 h,两相分离后超声脱气30 min。上相作固定相,下相作流动相,将固定相以20 mL/min的流速泵满管路,然后以850 r/min正转,流速2 mL/min的条件泵入流动相,温度25℃,检测波长280 nm。取20 mL下相溶解花色苷500 mg,进样20 mL后采集数据。根据高速逆流色谱峰峰型、保留率P和流出液情况判断分离效果。根据色谱分步收集 110~130 min、138~159 min、251~271 min、280~299 min 时的流出物,合并相同化合物的流出液,减压浓缩温度为40℃,冷冻干燥备用。取少量干粉甲醇溶解,经HPLC进样检测。
1.3.6 半制备高效液相色谱制备花色苷及其组分的条件优化
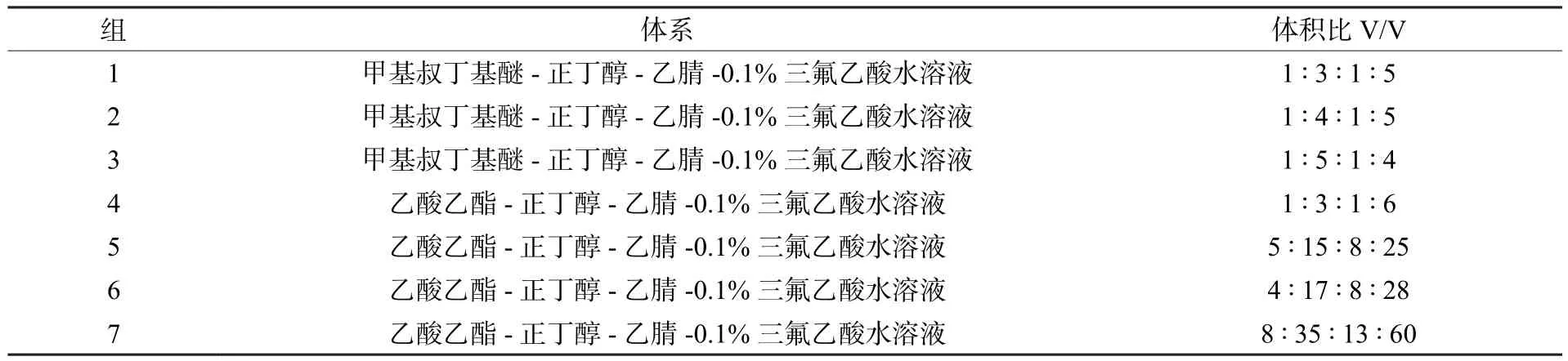
表1 不同溶剂体系设计方案Table 1 Designs of Different solvent systems
将高速逆流色谱法得到干粉用甲醇溶解,参考上述高效液相色谱条件,采用C18制备柱(20 mm×250 mm,10 μm),在波长 280 nm、柱温30℃的条件下,控制其他条件不变,分别探究不同流动相、不同流速、不同进样量、不同梯度洗脱方案对花色苷分离效果的影响[20],方案设计如表2,确定最佳色谱条件,根据色谱图分别收集不同组分,各组分收集浓缩后做二次进样分离。

表2 不同条件方案Table 2 Designs of Different condition scheme
1.3.7 花色苷含量分析方法
待检测样品液经0.45 μm有机滤膜过膜,高效液相色谱分析方法参照刘林峰等[21],每次进样10 μL,重复进样3次,按以下公式[7,22]计算样品液中花色苷含量。

式中:c为花色苷标准品浓度(μg / mL);m0为花色苷标准品质量(g);v0为标准品体积(mL);A1为标准品峰面积;A0为花色苷峰面积;ω为总花色苷质量分数(μg / g);A3为花色苷峰面积;v1为待测样品体积(mL);m1为待测样品质量(g);P 为花色苷得率(mg / g);m2为总花色苷质量。
2 结果与分析
2.1 萃取、脱脂、离子交换柱色谱与大孔树脂分离对花色苷得率的影响
萃取结果和离子色谱分离结果如表3。样品粗提的条件为避光,提取后得率5.64 mg/g;再经过有机相萃取脱脂处理、阳离子交换树脂层析后,得率提升;对树脂层析处理后的花色苷分析检测,其纯度为20.38%,说明其中含有较多的杂质。

表3 前处理工艺结果Table 3 Result of pretreatment processes
2.2 高速逆流色谱最佳溶剂体系的筛选
目标组分在两相溶剂体系中的分配系数K值[8]影响高速逆流色谱法的分离效果,K值偏大则出峰耗时过长,目标组分峰型变宽变矮,组分之间的分离度R较差;而K值偏小,各组分在固定相中的保留时间较短,出峰过快,导致流出组分难以分离。不同配比的溶剂体系的K值如表4所示,组别1、2、3、6中单个组分K值较大,组别4、5组分之间的K值相差较小,组分不能完全分离,筛选后确定组别7为最佳溶剂体系。
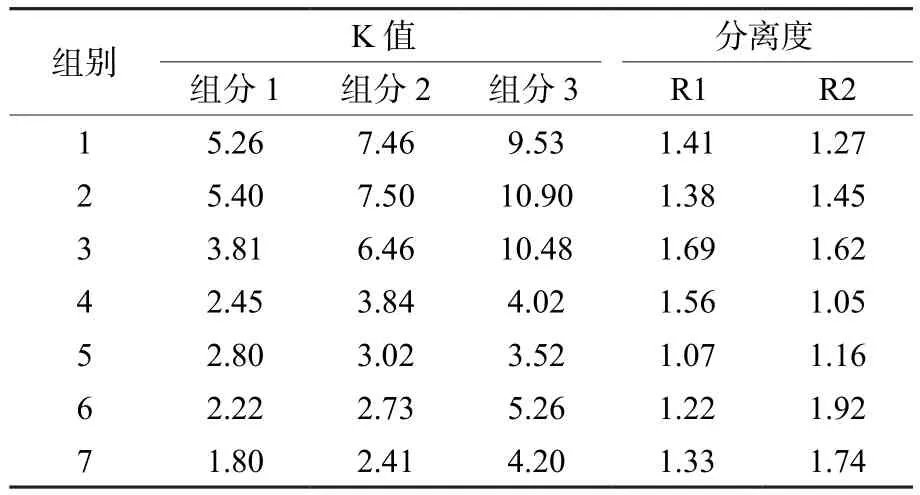
表4 不同溶剂体系的K值及分离度R值Table 4 K value and resolution R value in different solvent systems
在以乙酸乙酯-正丁醇-乙腈-0.1%三氟乙酸水(8∶35∶13∶60)为最佳的溶剂体系中,杂质峰与目标组分峰分离效果良好,保留率为56.8%,各组分在固定相中保留时间较长,可有效分离。收集流出液,经检测计算,以树脂吸附处理后的花色苷浓缩物为样品,经高速逆流色谱分离后,总花色苷得率达到160.59 mg/g。以高效液相色谱检测流出液,得到物Ⅰ和Ⅱ,物质Ⅰ主要是黄酮类物质,而物质Ⅱ主要是花色苷类物质,总花色苷纯度达到43.64%,将交换树脂层析后的粗品纯度提高一倍左右,由此可见,通过高速逆流色谱法可以分离出花色苷粗制品中黄酮类杂质,并有效提高花色苷粗制品纯度,适用于花色苷的纯化工艺。
2.3 半制备型高效液相色谱法条件优化
2.3.1 梯度洗脱筛选
通过调节各梯度的两相组成、梯度的变化速率,从而影响目标组分的分离效果。控制流动相为Ⅱ、流速为10 mL/min、进样量为1 mL,考察梯度洗脱方案的结果如图1所示,方案a出峰时间较晚,耗时较长;方案b、c出峰时间均提前,方案c出峰最早。相比于方案b的分离效果,方案c可实现各花色苷组分的有效分离,且实验消耗的流动相最少,故选择方案c用于后续研究。
2.3.2 洗脱流速筛选
在半制备高效液相色谱制备花色苷及组分过程中,流速会影响溶质、色谱柱和流动相之间的相互作用,进而导致目标组分保留时间的变化。一般来说,随着流速的持续增加,色谱分辨率有降低的趋势,而且过高的流速对于制备柱寿命的负面影响较大。控制流动相为Ⅱ、梯度洗脱方案为c、进样量为1 mL,考察流速方案的结果如图2所示,流速从5 mL/min增加到10 mL/min,出峰时间提前,耗时减小;随流速增大至20 mL/min,出峰时间进一步提前,但分离度明显下降,峰型变差,无法有效达到分离的目的,综合后选择5 mL/min的低流速作为较优的洗脱流速方案。

图1 不同洗脱程序方案分离花色苷的HPLC色谱图Fig. 1 HPLC Chromatograms of different elution procedure
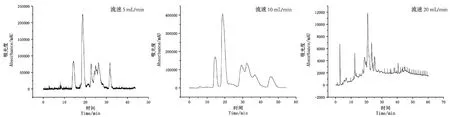
图2 不同洗脱流速分离花色苷的HPLC色谱图Fig. 2 HPLC Chromatograms of different elution fl ow rate
2.3.3 进样量筛选
适当增加单次进样量的优点在于节约时间,减少流动相消耗,但进样量过大,会出现柱压过载、分离度下降的问题。控制梯度洗脱方案为c、流动相为Ⅱ、流速为5 mL/min,考察进样量的结果如图3所示,进样量为0.5 mL时,分离效果较差,而进样量为2 mL时,出峰时间较晚,且分离度不如1 mL进样量时的分离效果,故选择1 mL为最优进样量方案。
2.3.4 流动相筛选
控制梯度洗脱方案为c、流速为5 mL/min、进样量为1 mL,以甲醇为有机相的方案Ⅰ不能将花色苷组分有效分离,方案Ⅱ出峰较早,且分离效果优于方案Ⅰ,而方案Ⅲ出峰时间虽较晚,但分离度较方案Ⅱ高,在保证分离度的前提下,综合三个方案,最终选择方案Ⅲ为最优流动相。
2.3.5 花色苷组分的制备
将高速逆流色谱纯化后的花色苷在甲醇中溶解,按上述筛选获得的最优色谱条件:C18柱(20×250 mm,10 μm)制备柱,波长 280 nm,流动相1%醋酸(A)-乙腈(B)、梯度洗脱(0~55 min,10%A~40%B),流速 5 m L/min,进样量为1 mL进样分离,根据出峰时间和峰形接收花色苷组分,各组分再浓缩二次进样分离。

图3 不同进样量方案分离花色苷的HPLC色谱图Fig. 3 HPLC Chromatograms of different injection volume

图4 制备型HPLC第二次分离花色苷组分的HPLC图Fig. 4 HPLC chromatogram ofAnthocyanin components
二次进样分离收取了5种花色苷组分流出液,各花色苷组分经分析型HPLC进样检测(图4),花色苷组分保留时间分别为14.8 min、23.1 min、28.4 min、31.3 min 和 33.6 min,纯度分别达到40.68%,24.38%,35.75%,90.61%和60.53%,其中组分4纯度最高。UV光谱图显示(图5)组分1、组分2、组分3、组分 4和组分 5分别在 523 nm、482 nm、534 nm、532 nm和520 nm处有最大吸收峰,而这5种组分在波长280 nm附近也存在最大吸收峰,符合花色苷类物质在可见光区500-540 nm附近和紫外光区280 nm附近存在最大吸收波长的特征[5,9,23],同时组分2在326 nm处存在一个吸收峰,研究发现[24,25]花青素分子在326-329 nm处存在的吸收峰是由于咖啡酸酯化的原因,故推测组分2为酰基化花色苷。
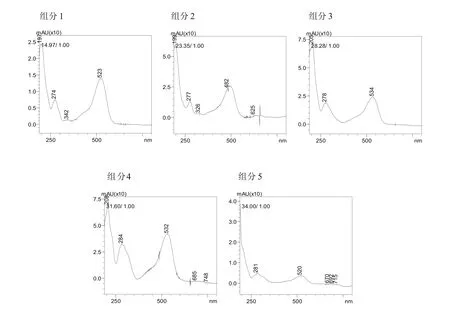
图5 制备型HPLC第二次分离花色苷组分的UV光谱图Fig. 5 UV spectrum of Anthocyanin components
3 结论与讨论
植物中花色苷本身的含量较低,而又因植物的种类不同,其含量存在差异,在茶树中也是如此。已有研究表明[26-27],茶树中含飞燕草素-3-O-β-半乳糖苷、矢车菊素-3-O-β-半乳糖苷、天竺葵素-3-芸香糖苷等多种花色苷组分,而各种组分分子量、糖苷位置等不同,所以花色苷组分的功能及分子极性也会相应地表现出差异,单一的分离技术可制备较高纯度的花色苷,但可能会因提取条件的限制而存在花色苷变质[12,28]、种类单一等问题,多种色谱方法结合并分离植物活成分性的研究也有报道[29],因此,从植物中制备花色苷需要协同多种分离技术,可以克服因植物原料中花色苷含量低、花色苷组分类型多、极性不同等造成制备花色苷的技术缺点,从而达到高效分离纯化植物中花色苷的目的。
本研究采用花色苷含量较高的紫芽茶树品种,对样品进行前处理,整个过程在避光、酸性条件下进行,获得花色苷的提取物,后续制备的浓缩液也及时冷冻干燥保存,保证在最初的原料及实验过程中,减少花色苷因变质的失损量,尽可能多的保留样品中的花色苷。进而以高速逆流色谱继续纯化花色苷,在该环节中,溶剂体系的优化极为重要,其决定了花色苷产物的最终得率。在本研究中,酸乙醇粗提物中得率仅为5.64 mg/g,但已高于文献报道的3.78 mg/g[12],而经过进一步处理,并以筛选出最佳的溶剂体系进行高速逆流色谱纯化除杂,得率达到160.59 mg/g,而纯度也由前处理的20.38%上升至43.64%,花色苷提取效率极大提升。最后采用半制备液相色谱,利用其分离纯度高、可制备量大、分离效率高等[20,30]等优点,分离高纯度的花色苷组分,通过优化的色谱条件,实验分离出5种花色苷组分,纯度最高达90%以上,说明半制备液相色谱对于紫芽茶花色苷的分离效率与采用的色谱条件具有重要的关系。综上所述,采用高速逆流色谱耦合半制备型液相色谱,分离纯化紫芽茶树花色苷的效率得到大幅提升,由实验结果可知,在对紫芽茶树花色苷及组分的分离中,高速逆流色谱结合半制备液相色谱是一种有效的分离制备花色苷的方法。
实验所分离的花色苷组分中纯度有高于90%的组分,但也有组分因为分离条件的限制而仅有20%以上,这样的纯度尚且达不到质谱鉴定的要求,其中组分2,根据文献可推测其为酰化花色苷,而这也需要对该组分深入的研究及分析,所以在花色苷分离纯化的后续研究中,还是要以分离纯化为基础,制备纯度更高的花色苷及其组分,协同多种工艺技术、把控关键环节及节点,深入分析在不同植物中花色苷类型、组分的存在方式及功能的差异性,从而真正达到制备高纯度花色苷的目的,发挥植物功能成分研究开发与利用的价值。
猜你喜欢
粉末冶金技术(2021年3期)2021-07-28
课堂内外(高中版)(2021年7期)2021-01-17
小学生作文(低年级适用)(2019年4期)2019-04-29
天然产物研究与开发(2018年5期)2018-06-13
散文诗(2017年18期)2018-01-31
红蜻蜓·低年级(2017年8期)2017-10-30
中学生数理化·高二版(2016年6期)2016-05-14
中国卫生(2015年6期)2015-01-22
天然产物研究与开发(2014年8期)2014-04-27
天然产物研究与开发(2014年6期)2014-04-27
