
江华苦茶的亲缘关系与遗传多样性研究
2019-04-06 11:56杨培迪罗军武
茶叶通讯 2019年2期
成 杨,刘 振,赵 洋,杨培迪,罗军武,杨 阳*
(1.湖南省农业科学院 茶叶研究所,国家茶树改良中心 湖南分中心,湖南 长沙 410125;2. 湖南农业大学 园艺园林学院 茶学系,湖南 长沙 410125)
江华苦茶是1987年湖南省认定的优良地方群体品种,主要分布在湖南省南岭山脉江华瑶族自治县,是宝贵的茶树资源[1]。DNA序列分析是区分个体间遗传差异最直接的方法,可提供基因组特异区的完全遗传信息,在植物的物种系统进化、分类和鉴定等方面起到了非常重要的作用[2-3]。作为DNA序列分析的一个部分,cpDNA序列有着相对保守、基因组较小、多拷贝、母系遗传的特点,被运用于各种层次的系统学研究[4-5],如被子植物基部类群[6]与物种系统进化研究[7]等,也有利用cpDNA进行山茶属植物的亲缘关系研究[8]。SSR分子标记具有通用性高、共显性遗传和多态性好等特点,是构建分子遗传连锁图谱、图位克隆、品种鉴定与指纹图谱、亲缘关系和遗传多样性研究以及辅助选择育种的有用工具,被广泛应用于茶树遗传多样性和群体遗传结构等研究[9-14],但是传统的SSR分子标记方法存在操作复杂、耗时耗力、不同批次数据整合困难、实验安全风险大等不足,给SSR标记在茶树中的应用带来一定困难[15-16]。
本研究在对江华苦茶进行调查的基础上,采用荧光SSR分子标记与cpDNA序列分析技术相结合的方法,对4个江华苦茶自然居群的32份资源进行了群体遗传多样性、遗传结构和遗传分化研究,探讨江华苦茶的亲缘关系、遗传多样性水平及群体遗传结构,为江华苦茶资源的收集、保护和研究提供理论依据。
1 材料和方法
1.1 材料
参试的32份茶树种质资源:江华瑶族自治县27份,其中码市镇龙湾村9份(MS)、蔚竹口乡12份(WZK)和大锡乡6份(DX),均为树龄较大、小乔木型的典型古茶树资源;湖南省茶叶研究所茶树种质资源圃苦茶资源5份(CYS)。实验材料均取一芽二叶新梢,液氮迅速冷冻处理,将其保存在-40℃冰箱中备用。
1.2 实验方法
1.2.1 引物合成
本研究采用3对cpDNA引物(表1),cpDNA序列为团队前期筛选的结果,由天根生化科技有限公司进行引物合成。15对SSR引物为团队已有的筛选结果,由上海生工公司合成,为了便于基因分型,在引物的5'端增加了一段序列(5'-CGTTGTAAAACGACGGCCAGT-3'),反应体系中增加了一个荧光标记引物(5'-FAMCGTTGTAAAACGACGGCCAGT-3')。
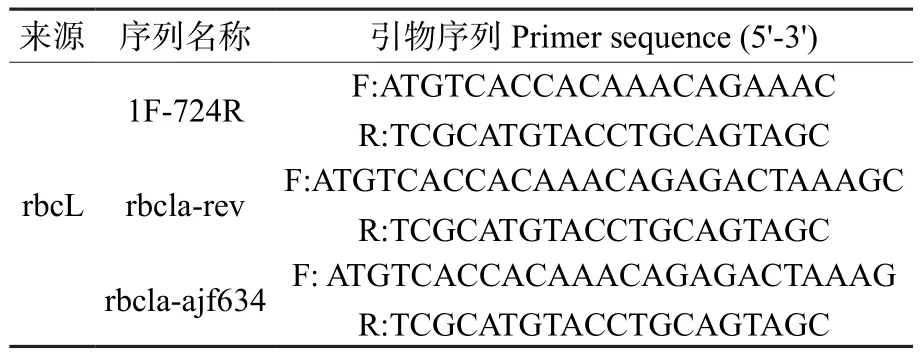
表1 cpDNA引物序列Table 1 cpDNA primer sequence
1.2.2 PCR扩增
cpDNA测序的扩增体系见表2、PCR反应程序见表3,扩增完成后,随机抽取扩增产物进行1.2%琼脂糖凝胶电泳检测,PCR产物送上海生工生物技术有限公司进行测序。SSR扩增体系见表4,采用降落式PCR,包括3个touchdown循环和后续的35个标准循环。起始变性温度为 94℃ 3 min,3个 touchdown循环为 94℃变性 30 s,68℃退火 1 min,每执行一个循环后退火温度降低2℃,72℃延伸1 min。35个标准循环为 94℃变性 30 s,58℃退火 45 s,72℃延伸45 s。完成以上循环以后,在72℃保持10 min,最后将PCR产物冷却到室温或者4℃。PCR扩 增 在Thermo-5020型PCR仪(Thermo Fisher Scienti fic. Int’l trade Co., Ltd.)上进行,扩增产物通过1.2%琼脂糖凝胶电泳检测,PCR 产物送上海生工生物技术有限公司进行基因分型。
1.2.3 数据处理
将cpDNA测序的结果采用DnaSP 5.1软件进行序列拼接,先剪去序列两端不可靠的碱基序列和引物序列,然后再将正反两个引物的序列进行比对,对序列进行编辑后获得正反引物序列一致的DNA序列。通过MEGA 6.0软件中的Clustal W 功能对所选材料的序列进行比对,适当手工校正,去除边缘不准确的部分序列,整理以待分析,计算变异碱基数[17]。采用MP法(most parsimonious tree, MP)构建分子系统树,对分支的可靠性评价使用靴带值(bootstrap,1000次重复)分析,自展数值75% ~ 100%表示强支持率,50%~74%表示弱支持率,< 50%表示不支持[18]。使用DnaSP 5.1软件对cpDNA序列进行单倍型数目(H)、单倍型多样性(Hd)、核苷酸多样性(π)、基因流(Nm) 、遗传分化系数(FST)等的计算,并进行 Tajima’s D、Fu and Li’s D*、Fu and Li's F* 的 中 性 检 验 (Neutrality tests)[19]及群体动态扩张的失配分布分析(Mismatch Distribution Analysis)。 采 用 Network 4.2.0.1软件基于最大简约性原则的中间连接网法分析(median-joining networks)构建不同单倍型间的网络关系图[20]。利用Arelequin 3.11软件中的 AMOVA( Analysis of Molecular Variance) 分析方法,基于cpDNA数据检测群体间和群体内的遗传变异[21]。

表2 cpDNA扩增体系Table 2 cpDNA ampli fication system

表3 PCR反应程序Table 3 PCR reaction procedure

表4 SSR扩增体系Table 4 SSR ampli fication system
使用 Popgen version 1.31 软件计算各 SSR 标记和居群的位点数(na)、有效位点数(ne)、Shannon信息指数(I)、期望杂合度(He)、观测杂合度(Ho)和Nei’s观测杂合度(Nei),计算居群间遗传一致度和遗传距离,并进行群体间的基因流(Nm)和F统计(F-Statistics)计算。采用ntsys 2.10e软件进行相似系数计算和聚类分析,利用Arelequin 3.11软件中的AMOVA( Analysis of Molecular Variance) 分析方法 ( Excof fier et al., 2005),基于 SSR 数据检测群体间和群体内的遗传变异。
2 结果分析
2.1 遗传多样性分析
2.1.1 cpDNA序列分析
3对引物测序在去除边缘不可靠部分后,分 别 获 得 634bp(1F-724R)、499 bp(rbclarev)、532 bp(rbcla-ajf634)的序列长度。序列的变异位点数量分别为5个、4个、4个。变异率从高到低分别是rbcla-rev(0.80%)、1F-724R(0.79%)和rbcla-ajf634(0.75%)。将3个序列对齐后依次拼接,共在4个居群中产生了5个单倍型(表5),单倍型数从多到少的居群依次为 WZK(4)、MS(3)、DX(3)和 CYS(2)。居群的Hd和π的大小顺序不一致(表7),Hd最大的为居群WZK,最小的为居群CYS;π最大的为居群MS,最小的为居群DX。江华苦茶群体单倍型多样性为Hd=0.726,核苷酸多样性为π=0.00233,群体遗传多样性相对较高。
2.1.2 SSR分子标记
15对SSR引物中扩增并分型成功的有13对(表6),共扩增等位位点99个,平均每对引物7.62个,位点数最多的为A05和A10(13个),最少的是A08(4个);有效位点最多的是A05(6.90个),最少的是A15(1.65个);Shannon信息指数最大的为A05(2.19),最小的是A15(0.82)。4个居群中(表6),平均na、ne、He、Ho、Nei最大的分别为5.23、3.13、0.62、0.65和0.60,最低的分别是3.54、2.48、0.50、0.59和0.54。遗传多样性最高的是蔚竹口乡居群(WZK),最低的是大锡乡群体(DX)。江华苦茶群体的He、Ho和Nei分别为0.56、0.65和0.64,群体具有较高的遗传多样性(表7)。
2.2 资源间的亲缘关系
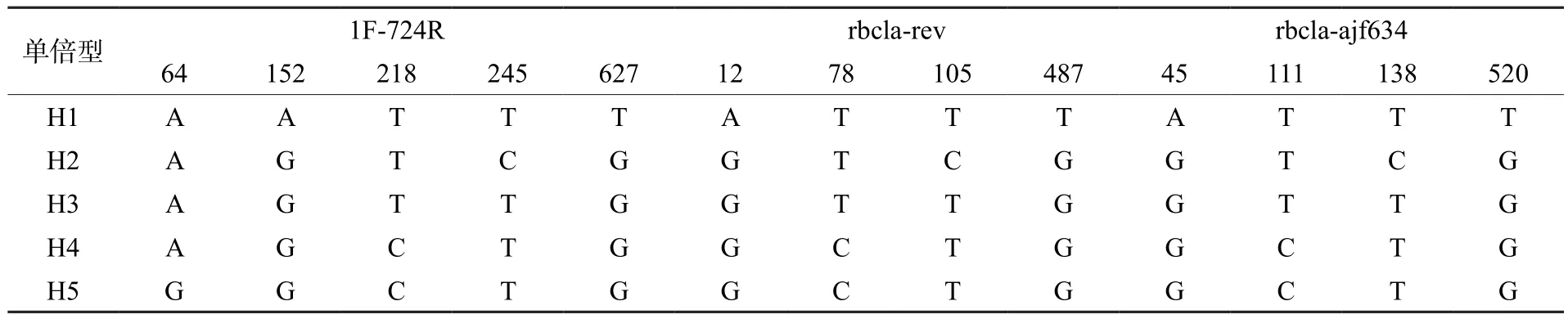
表5 3个cpDNA序列对齐产生5个单倍型的变异位点Table 5 Variable sites of the aligned sequences of three chloroplast DNA fragments in the 5 haplotypes (H1–H5)

表6 13对引物的扩增结果Table 6 13 ampli fication results of primers

表7 江华苦茶不同群体的遗传多样性Table 7 Genetic diversity of different populations of Jianghua bitter tea
利用MP法构建了江华苦茶32份资源的系统发育树(图1),结果表明,对照猪婆茶单独聚为一类(gene22),参试资源并没有依照地理类群聚为不同的小类群。
利用DnaSP 5.1软件对江华苦茶群体进行单倍型分析,分析结果表明,32个样品被分为了5种单倍型,各单倍型所包含的种质情况见表8。其中数量最多的单倍型为H3,包含13份资源;其次是H4,有9份资源。对比单倍型和分子系统树的结果可以发现,在系统树当中H5和H4单倍型聚在了一起,单倍型H5和H4的亲缘关系较近,而与其它单倍型的亲缘关系较远。

表8 基于cpDNA的江华苦茶单倍型的分布Table 8 Distribution of haplotypes of Jianghua bitter tea based on cpDNA
利用NETWORK构建叶绿体单倍型的网状进化图(图2),H3处于整个网络图的中心节点上,包含的资源数量最多。一般认为古老单倍型多位于网络图的内部节点,而新衍生出的单倍型一般位于外部节点,H3可能属于苦茶中比较原始的单倍型。以H3为基础,分别按照三条演化路线获得H1、H2、H4这三种单倍型,最后由H4演化出H5。

图1 基于三个序列和最大简约法(MP)构建的系统发育树(图中数字表示抽样检验的自展百分比)Fig. 1 Phylogenetic tree based on three sequences and maximum parsimony (MP) (the figure shows the percentage of selfexpansion of the sampling test)
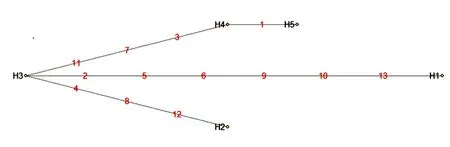
图2 cpDNA5种单倍型网状进化关系图Fig. 2 cpDNA 5 haplotype network evolution diagram

图3 ntsys对32个资源进行聚类分析构建的系统树Fig. 3 ntsys system tree constructed by cluster analysis of 32 resources
采用ntsys 2.10e软件对SSR分子标记数据进行处理,依照相似系数进行聚类分析见图3(cpDNA实验中22号的对照组在该实验当中没有使用,因此在该实验当中22号之后的资源排号均前进一位)。按照聚类图的结果,2号和14号、4号和13号相似性均高于0.95,而这两组在单倍型分析当中也属于同一单倍型,表明这两组资源的亲缘关系非常接近。而在32个样品当中,还有两个样品(3号和27号)与其他样品相似度均低于0.75,而在cpDNA的单倍型分析时,它们并没有与其他样品有明显差异,考虑到cpDNA的母系遗传特性,这两个样品的父本与其他样品存在比较大的差异性。在0.85以上的区间,存在一个集合紧密,数量众多的小集群,将该集群与cpDNA单倍型分析结合起来观察时,发现该集群包括了H3(4、8、13、16)、H4(17、20、21、32)和 H5(31)三个处于一条演化链上的单倍型,属于H3的四个资源相互之间关系最为密切,分别属于H4和H5的21号资源和31号资源的遗传相似度高达0.93,这个小集群是H3通过H4演化出H5在核基因上的体现。
2.3 居群遗传结构分析
采用13个SSR标记对所有的位点进行连锁不平衡检验,仅有3个位点是不连锁的(P>0.05)(表6),群体的近交系数FIS为正值(0.0344),而且一些位点显著偏离了Hardy-Weinberg平衡,表明群体的近交程度增加,纯合子比例升高。群体间的遗传分化系较小(FST=0.0971),分化程度较低,基因流较高(Nm=2.3254)。将SSR的检测结果分别进行AMOVA分析,结果表明江华苦茶的遗传变异主要出现在居群内(90.98%),居群间的遗传差异很小(表9)。

表9 SSR实验数据AMOVA分析结果(FST:0.09107)Table 9 AMOVA analysis results of SSR experimental data(FST: 0.09107)
利用DnaSP 5.1软件对四个居群的遗传结构分析表明,居群遗传分化系数FST=0.26019( P<0.0001),基因流Nm=0.71,居群间的遗传分化较大,基因流较弱。这个结果似乎表现了江华苦茶cpDNA的遗传变异主要存在于居群间,而对cpDNA实验数据采用AMOVA分析之后,得出的结果则是变异依然主要存在于居群内(表10),遗传分化并不明显。
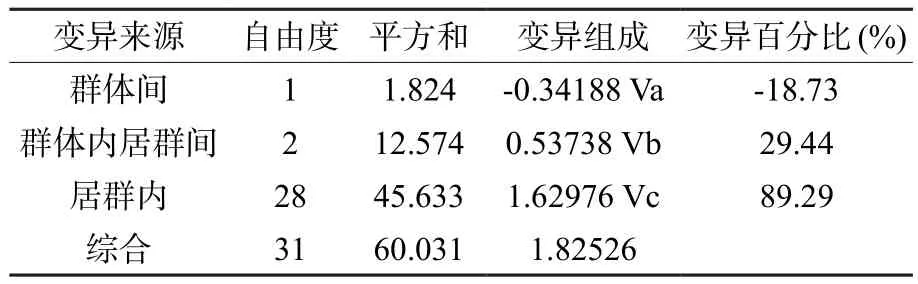
表10 cpDNA实验数据AMOVA分析结果(FST : 0.10710)Table 10 AMOVA analysis results of cpDNA experimental data (FST: 0.10710)
2.4 中性检验与失配分析
利用DnaSP 5.1软件对江华苦茶群体进行中性 检 验 ( Tajima,1989;Fuet al.,1993) , 检验结果表明:参试资源的 Tajima’s D、Fu and Li’s D 和 Fu and Li’s F 均为正值,统计检验也不存在显 著 性(Tajima's D: 0.65107,P> 0.10,Fu and Li’s D* test statistic: 1.04908,P> 0.10,Fu and Li’s F* test statistic: 1.08399,P> 0.10) 表 明 资源群体未经过扩张事件。对单倍型进行了失配分析,若群体经历了扩张或瓶颈效应,将导致失配分布曲线呈现单峰; 若群体处于动态平衡或缓慢衰退状态,没有经历扩张,则失配分布曲线为双峰或多峰 ( Tajima,1989;Fu et al.,1993);结果显示,失配分布曲线呈多峰曲线,表明江华苦茶群体均没有经历扩张(图4)。

图4 失配分布曲线Fig. 4 Mismatch distribution curve
3 讨论
3.1 江华苦茶群体遗传多样性
13对引物在32个江华苦茶资源中扩增的平均等位位点为7.62个,有效位点3.38,信息指数1.39,高于Zhao[22]等和Yao[23]等的研究结果,而与Fang[24]等和Wang[25]等的研究结果一致。这种丰富的等位位点与检测资源的遗传多样性有关,也与SSR扩增产物的检测技术等有关。本研究采用的荧光标记通过毛细管电泳进行检测,其分辨率要远高于传统的聚丙烯酰胺凝胶电泳技术,产生的等位位点就相对要多一些。
江华苦茶群体的Ho、He和Nei分别为0.56、0.65和0.64,高于Fang等和Yao 等的研究结果:Fang等采用58个SSR标记对185份中国茶树栽培品种的遗传多样性进行了研究,Ho平均为0.340;Yao等采用96个SSR标记对中国14个省份共450份茶树资源的研究,有13个省份的Nei小于本研究的0.64。cpDNA序列分析表明,江华苦茶群体的单倍型多样性和核苷酸多样性均较高,具有较高的遗传多样性。
3.2 江华苦茶群体亲缘关系
本研究对32种样品的cpDNA通过MP法构建了系统发育树,将样品分为了四个大类,大类内部自展百分比均在80%以上,获得了5种单倍型,对单倍型研究构建了网状分析进化图,发现了一个比较明显的cpDNA演化线路(H3-H4-H5)。nSSR标记的相似度研究32个样品之间的亲缘关系,两个样品(3号、27号)与其他样品相似度较远,以及1个聚类比较紧密的小集群(4、8、13、16、17、20、21、31、32),综合研究该集群内部相似度和样品在cpDNA单倍型当中位置能够很直观的体现出这些样品之间的亲缘关系。
3.3 群体遗传结构与基因流
基于SSR的江华苦茶群体的遗传分化水平(FST=0.0971),显著小于基于cpDNA的遗传分化(FST=0.26019 ),主要是因为核分子标记,基因流通过花粉和种子两种方式传播,而单亲遗传没有重组的cpDNA基因流只能通过种子流来传播,符合双亲遗传的核基因组分子标记更倾向于减弱群体间的遗传分化水平的观点。从实验数据结果来看,花粉流和种子流的比值要低于松属植物、川梨、珙桐等异交植物,表现出了较强的种子流。茶组植物为异花授粉且自交不亲和,江华群体的近交系数表现出正值(FIS=0.0344)不符合异交植物随机交配原则,表明江华苦茶在遗传演化过程中受到外来因素的影响,可能与人为的选择、运输和繁殖有关。
猜你喜欢
区域治理(2022年40期)2022-11-27
河北科技师范学院学报(2022年2期)2022-08-26
北京支部生活(2022年5期)2022-05-24
浙江中医药大学学报(2021年6期)2021-07-12
中国粮油学报(2020年12期)2021-01-09
动漫界·幼教365(小班)(2019年10期)2019-10-28
动漫界·幼教365(大班)(2019年10期)2019-10-28
动漫界·幼教365(中班)(2019年10期)2019-10-28
作文周刊·小学一年级版(2019年28期)2019-09-07
