
Radiation-induced malignant rhabdoid tumour of the hypothalamus in an adult: A case report
2019-03-21 13:13PeiMengNgPehHuehLowDonaldNgianSanLiewAlbertSiiHiengWong
World Journal of Clinical Oncology 2019年11期
Pei-Meng Ng, Peh-Hueh Low, Donald Ngian-San Liew, Albert Sii-Hieng Wong
Abstract BACKGROUND Rhabdoid tumours of the central nervous system are highly malignant and extremely rare in adults. To the best of our knowledge, only 87 cases of malignant rhabdoid tumour have been reported to date, inclusive of 4 cases with presumed radiation-induced aetiology. We report a case of malignant rhabdoid tumour in an adult with presumed radiation-induced aetiology to enrich the armamentarium of this disease entity, which may have some implications for early diagnosis and treatment of this rare disease in the future.CASE SUMMARY A 27-year-old male, who was exposed to cranial irradiation at the age of 4 years as part of the treatment for acute lymphoblastic leukaemia, presented with symptoms of raised intracranial pressure for one week. Brain magnetic resonance imaging revealed a heterogeneously enhancing lesion at the hypothalamus.Stereotactic biopsy was performed. Histopathological examination of the lesion showed malignant rhabdoid tumour. The disease progressed rapidly, with manifestation of leptomeningeal spread. He was started on craniospinal irradiation but treatment was suspended after 5.4 Gy, as he developed myelosuppression. His clinical condition deteriorated rapidly, and he succumbed to his illness within 2 mo.CONCLUSION This fifth case of radiation-induced central nervous system rhabdoid tumour reenforces the aggressive nature of this disease with poor prognosis.
Key words: Malignant rhabdoid tumour; Atypical teratoid rhabdoid tumour; Radiation induced malignancy; Case report
INTRODUCTION
The advancement in therapeutic measures in the oncology field has led to a significant improvement in the overall survival of cancer patients. Hence, we are facing more complications from those therapeutic measures, including an increase in the number of radiation-induced malignancies. Malignant rhabdoid tumour (MRT) of the central nervous system (CNS) in adult is very rare, and the literature available consists mainly of case reports or series. Despite the rarity of this disease entity, 4 cases[1-4]of presumed radiation-induced MRT in the brain have been reported to date. We present herein a case of hypothalamic tumour in an adult patient diagnosed with MRT following prophylactic irradiation for acute lymphoblastic leukaemia during childhood.
CASE PRESENTATION
Chief complaints
A 27-year-old male presented with headache, vomiting and blurring of vision for 1 wk duration.
History of present illness
The patient’s headache was of gradual onset, throbbing in nature, worse in the morning with associated projectile vomiting and blurring of vision. The headache was relieved temporarily by analgesia but progressively worsened over the course of a week.
History of past illness
The patient had history of acute lymphoblastic leukaemia at the age of four and was treated according to the Berlin-Frankfurt-Munster Protocol.
Physical examination
Neurological examination on the day of admission revealed bilateral reduction in visual acuity. Visual acuity for the right eye was light perception and for the left eye was 6/6. No other neurological deficit was noted. Systemic examinations were unremarkable.
Laboratory examinations
Blood investigation findings were normal.
Imaging examinations
Magnetic resonance imaging of the brain revealed a heterogeneously enhanced hypothalamic tumour (Figure 1). The lesion measured 2 cm × 1.5 cm, located mainly at the hypothalamus with extension into the optic chiasm. No pituitary stalk and pituitary gland involvement was detected.
Further diagnostic work-up
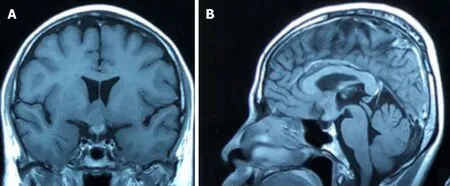
Figure 1 Magnetic resonance imaging of the brain, T1-weighted gadolinium-enhanced.
Stereotactic biopsy of the tumour was performed. Histopathological examination of the specimen showed focus of tumour infiltration that displayed rhabdoid morphology (Figure 2). Nuclei were eccentrically placed, hyperchromatic and pleomorphic with coarse chromatin. There was an ample amount of eosinophilic cytoplasm. Mitotic figures were seen. The cells stained positive for vimentin,cytokeratin (CK) AE1/AE3, glial fibrillary acidic protein (GFAP), cluster of differentiation (CD) 68 and CD138. Synaptophysin, human melanoma black 45, CK7,CK20, desmin, smooth muscle actin, CD79, CD30, terminal deoxynucleotidyl transferase, placental alkaline phosphatase, and leucocyte common antigen were negative upon staining. Ki67 was more than 20%.
FINAL DIAGNOSIS
The final diagnosis of the presented case was MRT of the CNS.
TREATMENT
The patient’s clinical condition deteriorated rapidly, with manifestation of leptomeningeal dissemination. He lost his vision within 5 d and developed multiple cranial neuropathies over the subsequent week. This started off with right eye ptosis,followed by bilateral lateral gaze palsy, loss of the left frontal furrow upon upward gaze with loss of the left nasolabial fold and drooping of the left angle of the mouth.His voice became softer, and he coughed upon swallowing both liquid and food. His tongue showed fasciculation with deviation to the right. This was followed by progressive bilateral lower limb weakness over the next week. The spinal magnetic resonance imaging showed leptomeningeal spread. Lumbar puncture was performed,and cerebrospinal fluid analysis revealed dispersed cells with rhabdoid features. The treatment plan was to administer 36 Gy of craniospinal radiation (20 fractions).However, treatment was halted after 5.4 Gy as he developed myelosuppression.
OUTCOME AND FOLLOW-UP
The patient’s general condition deteriorated rapidly after the radiotherapy was halted,and he succumbed to his illness within 2 mo from the onset of the illness.
DISCUSSION
MRTs of the brain were first described in 1987[5]. They have been characterised by diffuse growth of rhabdoid cells with typical paranuclear, glassy eosinophilic inclusions. Their nuclei are eccentrically placed, vesicular and with occasional prominent nucleoli, and mitosis is commonly found[6,7].
According to the World Health Organization classification of tumours of the CNS[8],MRT of the CNS with polyphenotypic (epithelial, mesenchymal, neuroendocrine)features by histology or immunohistochemical staining can be categorised into atypical teratoid rhabdoid tumour (ATRT) or CNS embryonal tumour with rhabdoid features based on their integrase interactor 1 (INI 1) status. In ATRT, there must be SMARCB1 (INI1) or SMARCA4 (BRG1) inactivation. Those with expression of SMARCB1 (INI1) and SMARCA4 (BRG1) or in which SMARCB1 and SMARCA4 status cannot be confirmed fall into the group of CNS embryonal tumour with rhabdoid features, despite having the same pathological features as ATRTs.
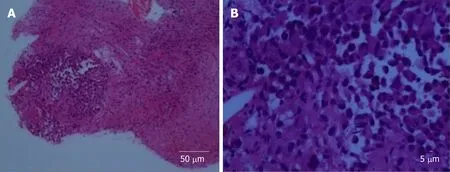
Figure 2 Histopathological examination.
Eighty-seven cases of MRT of the brain in adult has been reported,, with 39 of them located at midline and 47 cases off midline. The location was not mentioned in one case. For those located at the midline, 31 cases of sellar and suprasellar tumour were reported. Another 8 cases were located at the pineal region. Our case is the first in the literature to report an MRT located at the hypothalamus. This supports the hypothesis of Dardis et al[9], which states that the residual undifferentiated ectoderm in the circumventricular organ can be an origin for this tumour.
The appearances of rhabdoid tumour in the lung and ileum 20 years after radiation therapy (RT) for Wilms’ tumour have been reported by Litman et al[10], raising the possibilities of radiation-induced rhabdoid tumour. Four adult cases of presumed RTinduced MRT of the CNS have been reported in the literature[1-4]. The clinicopathological and imaging features of these cases and our case are summarised in Table 1, Table 2 and Table 3. Genetic analysis was performed in 1 of the 5 cases,which demonstrated loss of both SMARCB1 alleles at chromosome 22. Four cases showed inactivation of INI 1. The average age at the time of previous irradiation was 5.4 years, with a mean total radiation dose of 28 Gy and an average latency period of 23 years (Table 4). Overall prognosis is poorer compared to non-RT-induced MRT in adults, with a mean survival of 12 mo compared to 38 mo in the non-RT-induced group[1-4].
Imaging features for MRT of the CNS are non-specific. A combination of intratumoral haemorrhage, peripherally localised cysts, high cellularity seen as low T2 and apparent diffusion coefficient signal, as well as band-like or wavy enhancement can be seen. Around 15% of patients display meningeal dissemination at diagnosis[11]. For the 5 cases of presumed RT-induced MRT, all of them were located at the supratentorial compartment. Two cases were located at midline, and three cases were located off midline. Peritumoral oedema was more pronounced in the 3 cases located off midline, and peripheral tumour cysts were found in those 3 cases only. All cases demonstrated heterogenous contrast enhancement, with 3 of them showing the characteristic band-like enhancement. Leptomeningeal spread was present in 1 out of the 5 cases during the first presentation.
In the present case, the diagnosis of ATRT was suspected initially, but due to the rarity of this disease in adult this differential was deprioritized. A wide range of immunohistochemical stains were employed to exclude other possibilities first.Because of this, we found that our case was positive for CD138, which has never been reported for other cases in the past. CD138 is involved in molecular pathways that are dysregulated during carcinogenesis, and it can be an attractive target for immunotherapy in cancer treatment[12]. Other immunohistochemical stains that were positive in our case included vimentin, CKAE1/AE3, CD68, and GFAP. Staining for synaptophysin, human melanoma black 45, CK7, CK20, desmin, leucocyte common antigen, CD79, CD30, terminal deoxynucleotidyl transferase, and placental alkaline phosphatase were negative. Ki67 was increased (more than 20%).
GFAP staining in MRT of the CNS has been found to be strongly correlated with leptomeningeal spread and predicts a worse prognosis[9]. Our case further re-enforces this evidence, as the patient’s specimen was positive for GFAP staining and he manifested leptomeningeal spread early in the course of his disease with rapiddeterioration.
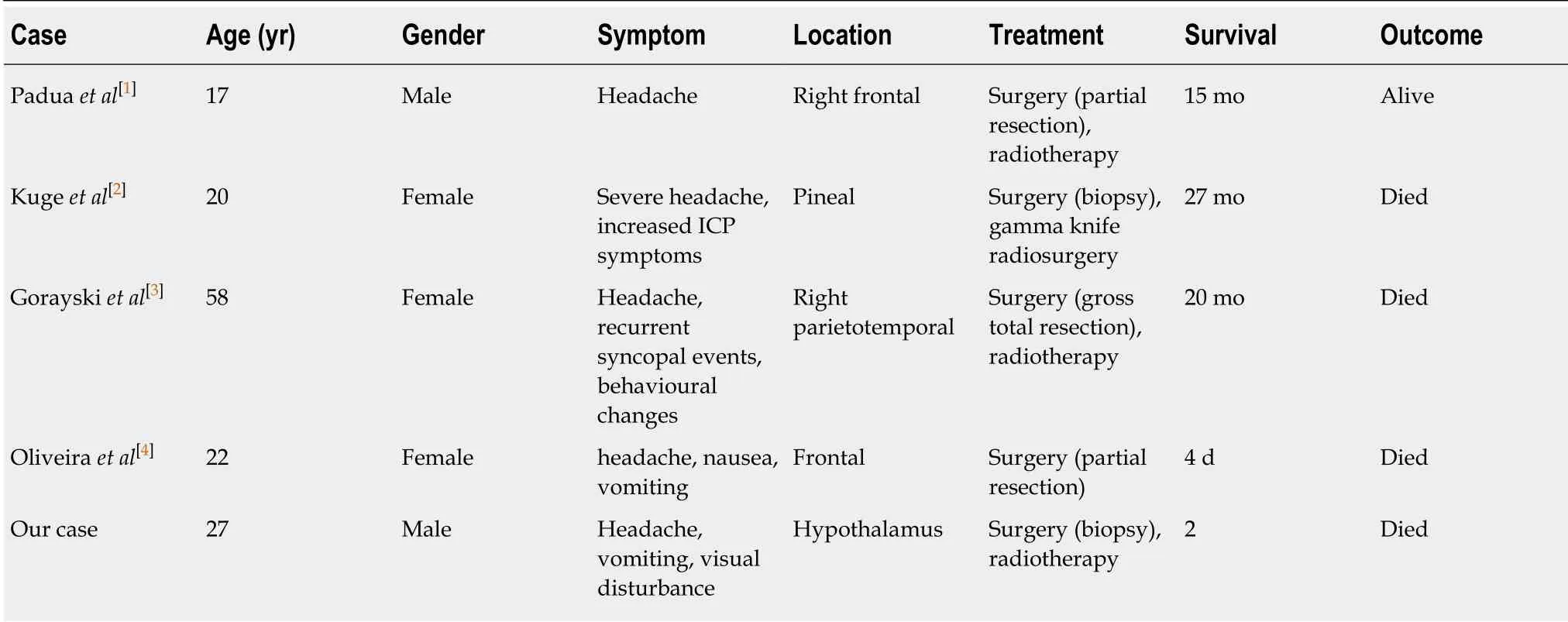
Table 1 Summary of patients, symptoms, treatments, and outcomes of radiation-induced malignant rhabdoid tumour
Multimodal therapy has been the proposed treatment strategy for MRT, by combining RT and chemotherapy following surgical resection[13,14]. Improved disease survival has been shown in patients given a total RT dose of more than 50 Gy[15].However, treatment strategies in the group of patients with a history of irradiation might need to be modified as re-irradiation can expose them to the risk of central nervous toxicity. The impact of toxicity depends on the dose, volume and time between exposures. Hence, re-irradiation in a patient with a history of RT requires tedious planning and good patient selection. Consideration should be given to other modes of treatment, such as stereotactic radiosurgery, which provide more precise and targeted tissue damage[16], or to local conformal proton therapy, which has shown encouraging results[17]. For candidates not suitable for re-irradiation, high-dose chemotherapy with autologous stem cell rescue may also be an option[18].
CONCLUSION
In summary, MRT of the CNS in adults remains a rare disease with an aggressive course and poor prognosis. Treatment recommendations for adults have been derived from paediatric data, due to the paucity of data available from adults. Despite being a dismal disease, prognosis in children has been more encouraging recently compared to 10 years ago, due to the continuous development and refinement of treatment strategies. We report a case of MRT in an adult with presumed radiation-induced aetiology, with the hope of enriching the armamentarium of this disease entity, which may have some implications for early diagnosis and treatment of this rare disease in the future.
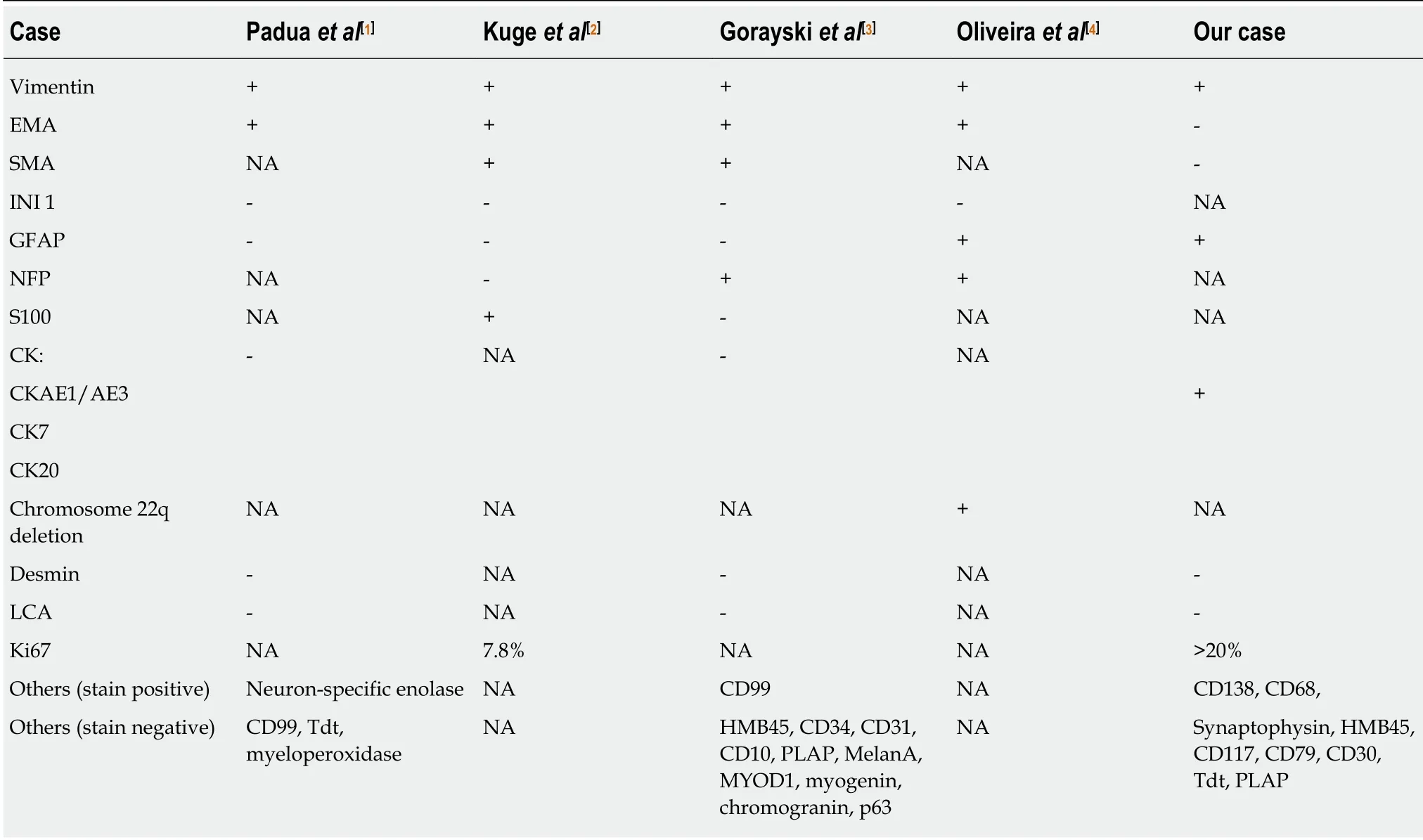
Table 2 Summary of immunohistological findings of radiation-induced malignant rhabdoid tumour

Table 3 Imaging features of radiation-induced malignant rhabdoid tumour
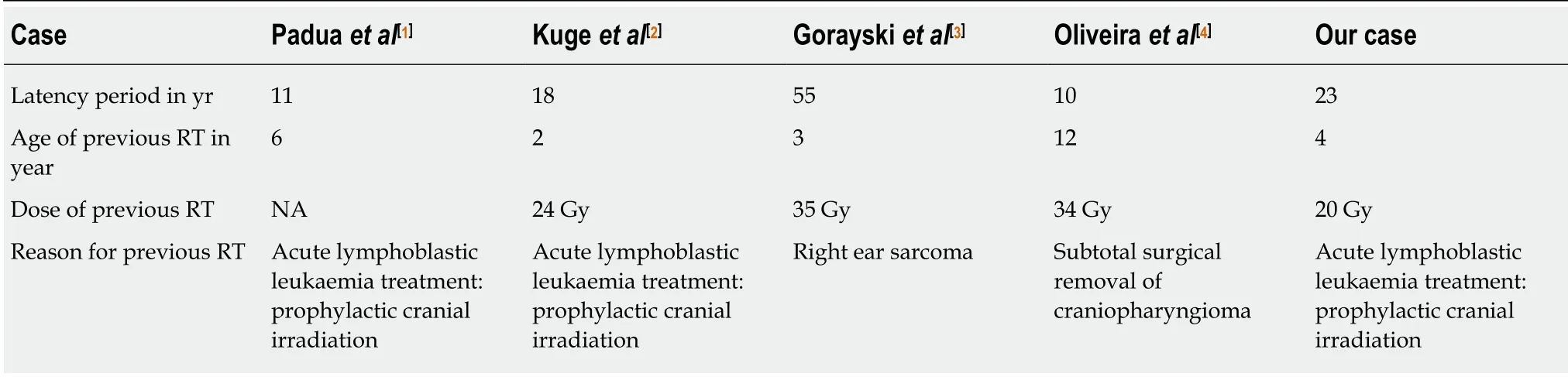
Table 4 Previous treatment details for radiation-induced malignant rhabdoid tumour
