
过渡金属Hf32-团簇的双重芳香性(σ与π)的理论研究
2019-03-19 09:21:14徐小俊池贤兴
原子与分子物理学报 2019年1期
徐小俊, 刘 勇, 池贤兴
(1.湖北第二师范学院 物理与电子信息学院, 武汉 430205; 2.温州大学 物理与电子信息学院,温州 325027)
1 引 言
芳香性最初是指象苯与它的衍生物这样一类重要的有机分子,它们具有特别的结构与化学上的稳定性[1-3].具有芳香性特征的团簇表现出强的磁屏蔽效应、大的环电流以及大的共振能等特别的性质,这些特性使其在非线性光学材料、导体、催化剂、吸附等方面有着潜在的应用.因此,对芳香性团簇进行量子化学计算,深入揭示分子团簇的电子结构和成键特征,不断加深对其组成-结构-性能关系的认识,可以为其潜在的应用打下坚实的理论基础.
近些年来,芳香性分子已经扩展到包括异原子体系[4, 5]与有机金属化合物[6, 7].最近,又发现了一些全金属原子团簇具有芳香性. Boldyrev 等人已经陆续报导了实验与理论上的证据证明全金属负离子团簇Al42-、Ga42-与In42-,异金属负离子团簇XAl3-(X=Si,Ge,Sn, Pb)也具有芳香性[7, 8],他们提出在中存在Al42-三重芳香性(一个π,二个不同的σ系统),在Al3-中存在着二重芳香性(一个π,一个σ系统).Huang等人[9]在理论与实验中指出,过渡金属团簇Mo3O92-和W3O92-的d轨道组成了σ芳香性. 近来,Boris B. Averkiev 与 Alexander I. Boldyrev理论上证明,过渡金属团簇Hf3具有三重芳香性(σ,πand δ)[10].
尽管到目前为止还没有一个完全满意的关于芳香性的定义,它们的物理本质是什么至今还有争论,但已有一些一般都接受的关于芳香性的判据,包括:环状平面等键长结构,能量高稳定性,非局域电子数满足Hückel (4n+2)规则,与芳香性磁特征等[11-13].近年来, Schleyer等人提出了一个新的芳香性磁判据,即核独立化学位移Nucleus-Independent Chemical Shift (NICS)[12]. NICS已对大量的芳香性化合物作了验证,发现是一个简单而十分有效的芳香性磁判据[13].
在本工作中,我们将运用两种密度泛函理论(B3LYP, B3PW91)和从头算方法MP2,对全金属团簇Hf32-的稳定结构与电子总能量(考虑了零点能ZPE)作理论计算, 以研究金属团簇Hf32-的芳香性.
2 计算方法
采用Guassian03W程序,运用两种密度泛函理论(B3LYP, B3PW91)和从头算方法MP2,分别对团簇Hf32-的线型D∞h和平面正三角形D3h结构进行优化. 对被选择优化的几何结构列在图1中.对Hf原子使用含有效核势近似(effective core potentials,ECPs)与相对论效应的LANL2DZ基组. 原子化能Atomazation energy(AE)采用了B3LYP和B3PW91两种方法,在优化得到的稳定的几何结构上,采用GIAO-B3LYP方法计算NICS[9]值.
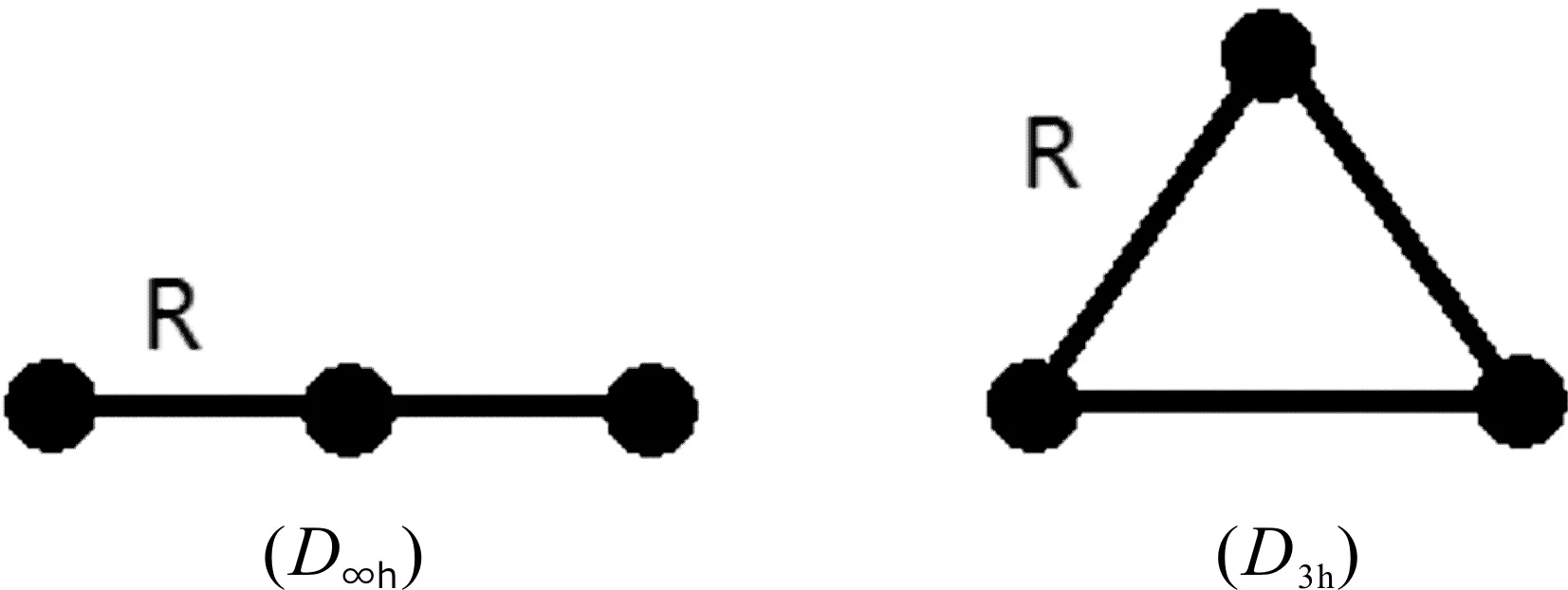
图1 全金属团簇Hf32-的优化结构Fig. 1 Theoptimized structures for all-metallic clusters Hf32-
表1 使用三种方法(B3LYP, B3PW91, Mp2)计算双金属团簇Hf32-的二种优化结构的键长Ri(Å)、总能量Etot和虚频数Nimag
Table 1 Optimized geometrical parameters (bond length R in A °), total electronic energies (Etotin Hartree) and number of imaginary frequencies (Nimag) of all-metallic clusters Hf32-with three methods (B3LYP, B3PW91, Mp2).

B3LYPB3PW91MP2D3hD∞hD3hD∞hD3hD∞hR2.74082.55072.72332.53482.91892.7107EreEtot-145.874053-145.791733-145.948852-145.854605-144.761005-144.527617Nimag020202υ1159(e)i40(πu)165(e)i51(πu)168(a1)i133(σu)υ2159(e)i40(πu)165(e)i51(πu)171(e)i95(πu)υ3211(a1)124(σg)219(a1)125(σg)171(e)i40(πu)υ4141(σu)149(σu)86(σg)
3 结果与讨论
3.1稳定结构
我们执行了Gaussian03w程序,全金属团簇Hf32-的线型D∞h和平面正三角形D3h结构进行优化,计算数据列在表 1.中. 计算结果显示,线性结构都有2个虚频,因此是不稳定的.而平面正三角形结构不存在虚频,因而是稳定结构.
3.2 芳香性能量特征
原子化能(AE)是一种比较好的芳香性能量判据,本文对双金属团簇Hf32-原子化能计算如下:

从表中可以看出计算得到的芳香性稳定化能(AE)均为较大值,表明该团簇具有芳香性.
表2 Hf32-的原子化能(单位:kJ/mol)
Table 2 Atomization energies (kJ/mol)for all-metallic clusters Hf32-

B3LYPB3PW91MP2Hf32-55.7101227
3.3 磁性质
NICS是independent chemical shifts的缩写, NICS负值越大,则芳香性越强.本文取6种全金属团簇Hf32-平面D3h结构的四个位置来计算NICS:一个在对称中心,另三个在中心上方分别离中心0.5 Å、1.0 Å、1.5 Å处.从表3中可看到, NICS值均小于零,而且从中心往上一直逐渐减小.对团簇的磁性质的分析指出, 全金属团簇Hf32-平面D3h结构具有芳香性..

表3 计算的NICS值 (ppm cgsu).
3.4 分子轨道分析
Hf原子的价电子组态为:5d26s2, 因此全金属团簇Hf32-(1A1’),共有14个价电子,占据着7个价分子轨道,分别为HOMO, HOMO-n(n=1-6). 通过仔细的布居分析,HOMO-n(n=1-15)主要是由Hf原子的s, d轨道组成.Homo-2(1a2")是离域化的π-MOS,每一个轨道都是由三个垂直于分子平面的d轨道叠加而成,而这些d轨道是主要由Hf原子的dxz和dyz原子轨道径向分子平面中心组成.Homo-3(2a1’)可看作是离域化的σ-MOS,每一个轨道都是由三个位于分子平面内的d轨道叠加而成,而这些d轨道是主要由Hf原子的dx2-y2和dxy原子轨道径向分子平面中心组成. 剩下的四个价分子轨道:Hf32-(1A1’)团簇的HOMO (1e'),Homo-1(1e’’), Homo-4(1e’) 和Homo-5(1e’), 它们分别是成键,反键和非键,且主要都是由Hf原子满壳层的s轨道组成的,对成键总的效果可看作为零.所以2个离域化的σ电子, 2个离域化的π电子,分别遵循4n+2电子计算规则,并且呈现出σ与π双重芳香性.
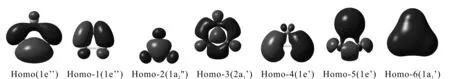
图2 全金属团簇Hf32-(1A1’)的价分子轨道图Fig. 2 Valence molecular orbital pictures for dianions Hf32-.
4 结 论
对全金属团簇Hf32-的稳定结构、虚频数,原子化能和电子总能量进了分析和计算. 全金属团簇Hf32-仅有一种稳定结构平面D3h结构. 在得到的稳定结构的基础上,计算了金属团簇D3h的磁性质:核独立化学位移(NICS),结果显示,所有的NICS值都是较大负值,这说明了存在较强的芳香性. 最后由分子轨道分析得出,全金属团簇Hf32-呈现σ与π双重芳香性.
猜你喜欢
合成树脂及塑料(2022年4期)2023-01-05 13:57:07
大学化学(2021年8期)2021-09-26 10:50:46
科技创新导报(2021年3期)2021-07-28 05:01:43
合成树脂及塑料(2021年6期)2021-01-09 01:56:30
山东化工(2020年5期)2020-04-07 09:59:30
天津行政学院学报(2019年4期)2019-10-08 09:17:44
中外书摘(2017年2期)2017-02-10 19:42:15
湖南有色金属(2016年3期)2016-05-18 03:00:19
电脑爱好者(2015年19期)2015-09-10 01:10:55
社会科学研究(2014年4期)2014-08-16 16:47:32
