
注射用头孢呋辛钠杂质谱研究
2019-03-13 07:07:44邓贵福
中国抗生素杂志 2019年2期
邓贵福
(1 重庆市食品药品检验检测研究院,重庆 401121;2 重庆市药物过程与质量控制工程技术研究中心,重庆 401121;3 重庆市化学药品质量控制与评价协同创新中心,重庆 401121)
头孢呋辛(cefuroxime)是英国葛兰素公司于1974年研究开发的第二代头孢菌素类抗生素,抗菌活性强,临床用于敏感的致病菌引起呼吸道感染、耳鼻喉感染、泌尿系统感染、败血症、脑膜炎、骨和关节感染、皮肤软组织感染、腹腔感染、生殖系统感染包括淋病,也可用于术前预防感染[1]。不良反应较少见,多表现为轻度过敏反应和消化道症状等[2]。上市剂型主要为粉针剂,美国有粉液双室袋产品上市。
头孢菌素一般由微生物发酵、纯化、精制以及化学修饰等过程制得,可能含有的杂质情况较常规化学合成药品更为复杂[3]。此外,头孢菌素结构不稳定,在生产及储存过程中遇光、热、水等条件均会发生降解反应,形成一系列的降解物及聚合物[4],使其失去抗菌活性,甚至产生毒副反应。因此,对药品的杂质谱进行研究,明确杂质结构和产生机理,对改进生产工艺,优化储存条件,提高产品质量具有重要意义。
中国药典2015年版已收载头孢呋辛钠原料及注射用头孢呋辛钠,头孢呋辛钠原料药还收载于美国药典40版、英国药典2017版及欧洲药典8.0版。其中美国药典40版未对有关物质进行控制,中国药典仅控制了单个杂质和总杂质[5],未对头孢呋辛钠的特定杂质(工艺或降解杂质)进行针对性的控制;欧洲药典8.0版提供了9个杂质的结构,并规定去氨甲酰头孢呋辛的含量不得过1.0%,还规定了单个杂质和总杂质的限度[6]。
本研究对中国药典和欧洲药典收载的有关物质进行了对比研究,结果显示,中国药典方法检出的杂质个数更多,杂质含量更高,且由于采用的梯度洗脱方式,各杂质的分布较均匀,分离度更好。后续研究均采用中国药典方法对注射用头孢呋辛钠的有关物质进行研究,并采用多个已知杂质进行了方法学验证。
本研究对国内两个厂家的注射用头孢呋辛钠和原研品“西力欣”的杂质谱进行了对比分析,并对检出的主要杂质进行了结构解析和来源分析,为完善药品质量标准,保证临床用药的安全性提供了参考。
1 仪器和试药
1.1 仪器
Waters高效液相色谱仪(Waters e2695,PDA检测器)。色谱柱:Agilent Eclipse Plus C8(5μm,4.6mm×250mm)。布鲁克Impact II飞行时间质谱仪,电喷雾电离源(ESI)。AX205分析天平(瑞士万通)。
1.2 试药
乙腈为色谱纯,水为超纯水,醋酸钠和冰醋酸等其他试剂为分析纯。注射用头孢呋辛钠A厂家(批号:17011614)和B厂家(批号:20170701)均为0.75g,原研品“西力欣”(注射用头孢呋辛钠,0.75g规格,批号:J853,葛兰素制药公司生产)。杂质对照品A(批号:17-04-1901,纯度95.3%)、E(批号:17-05-2005,纯度95.9%)、H(批号:17-04-2003,纯度98.7%)由Sinco pharmachem公司提供,B(批号:3-NAV-50-1,纯度97.0%)、C(批号:7-FLI-89-1,纯度96.0%)、D(批号:17-04-2002,纯度95.5%)、F(批号:21-MAY-17-12,纯度97.2%)、G(批号:2464-065A10,纯度96.8%)、I(批号:2-MSW-107-1,纯度97.0%)杂质对照品由LGC公司提供,头孢呋辛对照品(批号:130493-201105,含量92.1%)由中国食品药品检定研究院提供。
2 方法与结果
2.1 色谱条件与系统适用性试验
照《中国药典》2015年版二部“注射用头孢呋辛钠”项下的有关物质检查方法进行测定。
2.2 有关物质方法学验证
2.2.1 专属性
分别称取头孢呋辛及各杂质对照品适量,分别配制成浓度约为0.1mg/mL的储备液,再稀释为0.025mg/mL的溶液,进样,进行各组分的定位。同时配制各组分的混合溶液,记录色谱图,结果显示各相邻峰的分离度均大于1.5,说明本方法的专属性良好。
图中已积分的各峰依次分别为杂质I和A、头孢呋辛、杂质F、E、C、B、H、G及杂质D(图1)。
2.2.2 强制降解试验
取供试品适量在水解(60℃, 30min)、碱降解(1mol/L的氢氧化钠溶液)、酸降解(1mol/L的盐酸溶液)、氧化降解(1%的H2O2溶液)、高温(105℃, 16h)、光照[(4500±500)Lx, 4d]条件下破坏后进行测定,结果显示各破坏条件下主峰与杂质能有效分离。各破坏条件下本品的降解杂质产生情况见表1。
2.2.3 定量限和检测限
取头孢呋辛和各杂质对照品的储备液,采用逐级稀释法制备稀释溶液,进样20μL,取相当于基线噪声10倍时溶液浓度做为最低定量限,取相当于基线噪声3倍时溶液浓度做为最低检测限,记录色谱图。经计算,杂质I的最低定量限约为主成分的0.007%,最低检测限约为主成分的0.002%;杂质A的最低定量限约为主成分的0.04%,最低检测限约为主成分的0.01%;其他组分的最低定量限约为主成分的0.07%,最低检测限约为主成分的0.02%。
2.3.4 其他项目
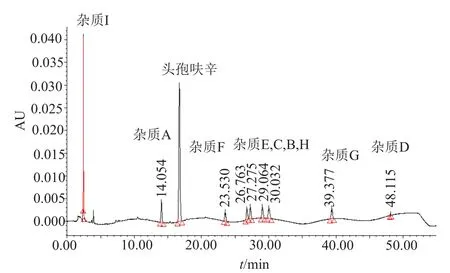
图1 杂质混合溶液色谱图Fig. 1 The chromatogram of impurity mixed solution
系统适用性、线性与范围、准确度、重复性、中间精密度等均满足《药品质量标准分析方法验证指导原则》的要求。

表1 头孢呋辛各杂质降解途径分析Tab. 1 The impurities degradation pathway of cefuroxime sodium
2.4 杂质测定结果
分别配制国内两个厂家产品和原研品的供试品溶液,进样进行有关物质测定,结果见表2。
由表2可知,同原研品相比,国内两个厂家的杂质个数较原研品稍少,但是杂质的总量稍大。不同来源样品中检出的主要杂质(接近或超过0.1%)相同,分别为相对保留时间为0.86、1.68和1.84的杂质。
2.5 杂质定性研究

表2 头孢呋辛钠杂质谱比较Tab. 2 Comparison of the impurity profiles of cefuroxime sodium
为明确各样品中检出主要杂质的结构和来源,本研究改进了液相色谱条件(将有关物质方法中的流动相A改变为5mmol/L的乙酸铵溶液,用甲酸调节pH至3.4,以与质谱体系兼容,其他条件不变),以使色谱流出物用于质谱分析。
质谱分析条件:离子化方式:ESI(+);数据采集范围m/z50~600;雾化气压力:0.4Bar,干燥气温度:180℃,干燥气流速:4L/min,碰撞电压:7eV;毛细管电压:4kV,锥孔电压:2kV。
3个厂家的产品中均出现了3个较大的杂质(相对于主峰的保留时间分别为0.86、1.68和1.84),其中国内两个厂家这3个杂质均超过了0.1%,分别与已知杂质A、E、H的相对保留时间基本一致,为进一步确认杂质的结构,本研究采用LC-MS对这3个杂质峰的截留物和已知杂质A、E、H进行了对比分析,相对保留时间为0.86杂质的分子离子峰[M+H]+的质量数质荷比为382.0666,与杂质A的质量数相符;相对保留时间为1.68杂质的分子离子峰[M+Na]+的质量数质荷比为447.0562,与杂质E的质量数相符;相对保留时间为1.84杂质的分子离子峰[M+H]+的质量数质荷比为364.0583,与杂质H的质量数相符;同时对各杂质进行了二级质谱分析,相对于主峰的保留时间分别为0.86、1.68和1.84的3个杂质的一级、二级碎片峰分别与已知杂质A、E、H质谱图一致,可以推定为对应的已知杂质。各杂质的一级、二级质谱图,以及相应的裂解途径见图2~4。
3 讨论
不同来源产品中检出的3个较大杂质分别为杂质A(去氨甲酰头孢呋辛),为头孢呋辛钠在酸性等条件下水解脱去甲酰胺基团而得;杂质E(头孢呋辛反式异构体)为头孢呋辛钠原料药合成过程中的工艺杂质,也可能由于头孢呋辛钠在存放过程中结构发生反转而得到的反式异构体;杂质H(头孢呋辛内酯),为头孢呋辛或去氨甲酰头孢呋辛在存放过程中由于高温或光照等的影响而发生分子内酯化反应而得的内酯物;上述3个杂质均可由头孢呋辛钠降解产生,需要有针对性地制定科学的限度,以更好控制产品质量。
目前中国药典中有关物质方法对杂质的分离能力和检出能力均较好,适用于本品的有关物质检测,但是其控制单个杂质的限度为1.0,总杂质的限度为3.0%。尚不满足目前药品技术审评审批的技术要求,建议将杂质A、E、H作为已知杂质进行控制(本研究采用杂质对照品进行了校正因子测定,杂质A、E、H相对于主成分校正因子分别为0.91、1.09和1.00,可以采用不加校正因子的主成分自身对照法计算杂质含量),限度订为1.0%,同时增加对未知杂质的控制,限度参照EMA抗生素有关物质标准的指导原则[7],订为0.2%,总杂质的限度仍为3.0%。
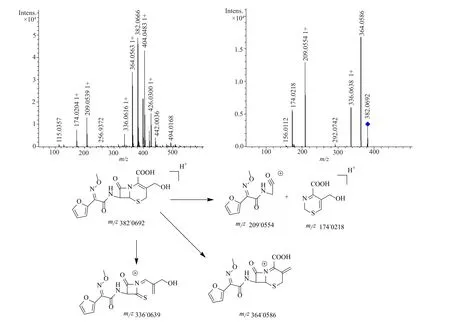
图2 杂质A的一级、二级质谱图及可能的裂解途径Fig. 2 MS and MS2 spectrum of impurity A and fragmentation pathway of impurity A
本文对中国药典2015年版注射用头孢呋辛钠有关物质方法对杂质的分离和检出能力进行了验证,结果表明适用于注射用头孢呋辛钠的有关物质检测;对比分析了两个国内厂家产品和原研产品的杂质谱情况,结果显示杂质个数和杂质含量没有显著性差别,并对样品中检出的较大杂质进行了定性分析;同时对《中国药典》2015年版注射用头孢呋辛钠的有关物质限度控制给出了合理的建议,为更好地保证注射用头孢呋辛钠产品质量提供了参考。

图3 杂质E的一级、二级质谱图及可能的裂解途径Fig. 3 MS and MS2 spectrum of impurity E and fragmentation pathway of impurity E
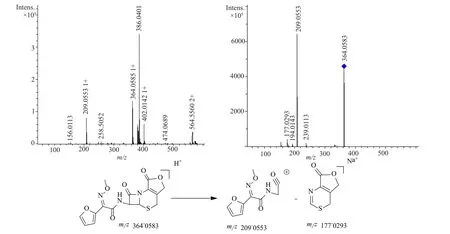
图4 杂质H的一级、二级质谱图及可能的裂解途径Fig. 4 MS and MS2 spectrum of impurity H and fragmentation pathway of impurity H
猜你喜欢
云南医药(2021年3期)2021-07-21 05:40:38
中华养生保健(2020年9期)2021-01-18 03:11:48
药品评价(2020年13期)2020-10-14 03:50:40
工业设计(2020年6期)2020-07-30 14:05:39
临床医药文献杂志(电子版)(2020年28期)2020-02-28 05:53:27
中国生殖健康(2019年2期)2019-08-23 08:11:54
国外医药(抗生素分册)(2016年2期)2016-07-12 14:24:59
中国卫生标准管理(2015年7期)2016-01-15 03:58:40
中国药业(2014年12期)2014-06-06 02:17:26
中国药业(2014年24期)2014-05-26 09:00:28
