
多基团功能化氧化石墨烯协同催化CO2环加成反应
2019-03-13 03:07兰东辉代威力谭年元伍水生区泽堂
无机化学学报 2019年3期
兰东辉 唐 婷 代威力 谭年元 伍水生 区泽堂*, 易 兵*,
(1湖南工程学院化学化工学院,环境催化与废弃物再生化湖南省重点实验室,湘潭 431000)
(2南昌航空大学,江西省持久性污染物控制与资源循环利用重点实验室,南昌 330063)
0 引 言
莫纳罗亚天文台测量结果显示:2017年5月大气中CO2的平均浓度远高于工业革命之前(0.028%(V/V)),达0.041%(V/V),创历史新高。大气中CO2浓度逐年增加导致温室效应加剧,这严重威胁到人类的生存和发展。直接以废气中的CO2为C1资源合成高附加值的化工产品,可避免CO2捕集与封存过程中的能耗高和潜在风险大等问题,满足绿色发展的要求,有利于改善生态环境,建设美丽中国[1]。
以CO2和环氧化物环加成反应合成具有高附加值的环状碳酸酯,不仅符合“绿色化学”的原则,而且满足“可持续性发展”的战略要求,是一种一举两得的技术[2-3]。新型高效催化剂的研制是CO2环加成反应合成环状碳酸酯工艺研究的重点[4-5]。其中离子液体(ILs)由于具有绿色环保、良好的溶解能力、催化活性高、结构和性能易调变等特点,被广泛用于CO2环加成反应[6-7]。张锁江和韩布兴院士等从电子、分子、团簇不同尺度出发设计并制备了一系列的功能化ILs,利用密度泛函理论证明羟基和羧基等氢键给体与卤素阴离子协同作用能促进环氧化物开环活化,降低CO2环加成反应的活化能[8-9]。但ILs的分离问题限制了其在环加成反应中的应用,将ILs固载是解决其分离问题的有效方法之一。
氧化石墨烯(GO)由于具有比表面积高、稳定性优异以及丰富的表面基团易于被功能化等特点,在无金属多相催化剂领域的应用引起了研究者广泛的关注[10-11]。最近,Yin课题组[12]和 Li课题组[13]利用与硅基材料负载ILs类似的方法研制了ILs功能化的GO,发现氢键给体和卤素阴离子协同催化作用有利于环氧化物的开环活化。但上述催化剂固载量不高,导致催化CO2环加成反应条件较苛刻(温度一般高于120℃),且ILs前驱体价格较贵。为解决ILs负载量受GO表面羟基数目的限制,本文提出多阳离子和多功能化的策略,基于氢键给体、卤素阴离子和胺协同催化CO2环加成反应的设计思想,以富含氮和三级胺且廉价的六次甲基四胺(Hatm)为前驱体合成羟基、季铵盐和叔胺功能化的GO,随后利用卤代烃进一步季铵化构建多基团和多阳离子ILs功能化的GO,并将其应用于催化CO2环加成反应。
1 实验部分
1.1 材料与试剂
Hatm、NaI、3-氯丙基三甲氧基硅烷、N,N-二甲基甲酰胺(DMF)、正溴丁烷(n-BuBr)、甲苯、三乙胺、三乙烯二胺、四甲基乙二胺、联苯和环氧丙烷均为分析纯,使用前未经过处理。
1.2 催化剂的制备
催化剂的制备过程如下:将2.2 mmol Hatm、2.7 mmol NaI、2.2 mmol 3-氯丙基三甲氧基硅烷和20 mL DMF依次加入到100 mL反应管中,随后密封置于80℃的油浴锅内搅拌反应20 h;上述反应液冷却后,在N2保护下,加入一定量的正溴丁烷(n-BuBr)和10.7 mmol NaI,继续80℃避光搅拌反应20 h;上述反应液冷却后,加入0.2 g GO和20 mL甲苯超声后于80℃搅拌反应20 h;反应结束后依次用乙醇离心洗涤6次,水洗3次,乙醇洗涤1次,60℃下真空干燥 20 h。依据加入 n-BuBr的量(0、2.2、4.4、8.8 mmol),分别将制得的催化剂依次记为GO-H、GO-H-Bu1、GO-H-Bu2和 GO-H-Bu4。 采用相同的方法,分别以三乙胺、三乙烯二胺和四甲基乙二胺代替Hatm为三级胺前驱体,不加入n-BuBr制得的催化剂依次记为GO-TEA、GO-D和GO-T。此外,分别以三乙烯二胺和四甲基乙二胺为前驱体和2.2 mmol n-BuBr为季铵化试剂制得的催化剂分别记为GO-D-B1和 GO-T-B1。
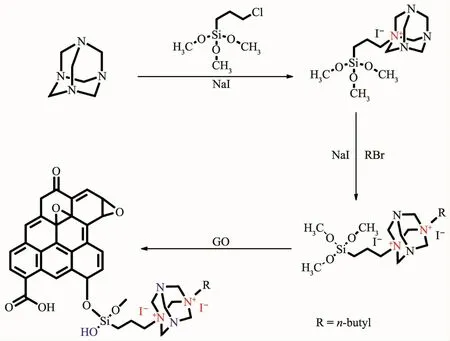
图1 GO-H-Bu4的制备过程Fig.1 Preparation process for GO-H-Bu4
1.3 催化剂的表征
傅里叶红外光谱仪 (FT-IR)分析了GO、GO-HBu1和GO-H-Bu4表面的特征基团,利用KBr压片法,采用德国布鲁克光谱仪器公司生产的Vector 22型仪傅里叶红外光谱仪进行表征。在美国热电集团公司Flash 2000元素分析仪(EA)上分析材料的组成。采用美国麦克比表面积测试仪(BET)分析了GOH-Bu4的比表面积和孔结构。GO、GO-H和GO-HBu4材料的表面组成使用赛默飞世尔科技公司KAlpha 1063型X射线光电子能谱仪 (XPS)进行分析,铝Kα微聚集单色器,电压和电流分别为12 kV和6 mA。热重分析(TG)在德国NETZSCH-STA-449C型热重分析仪上进行测试,分析条件如下:氮气气氛,加热范围为室温至550℃,升温速率为5℃·min-1。
1.4 催化剂活性评价
在30 mL间歇反应釜中加入一定量的催化剂和28.6 mmol环氧丙烷(PO);随后向釜内充入一定压力的CO2;置于所需反应温度下加热反应一定时间。反应完成后,采用冰水混合物将釜冷至0℃,放出剩余气体后加入联苯作内标,利用Agilent Technologies 7820A气相色谱进行分析。
2 结果与讨论
2.1 催化剂的表征
图2为 GO、GO-H-Bu1和 GO-H-Bu4的 FT-IR 谱图。与GO的红外谱图对比,GO-H-Bu1和GO-H-Bu4的谱图中出现了季铵 N(1 216 cm-1)、Si-O-C(727 cm-1)、Si-OH(909 和 3 417 cm-1)的振动峰[12-13]。 另外,GO经过功能化后C-O-C和C-OH峰减弱,证明GO表面含氧官能团减少,Zhang等[14]报道GO表面的羟基和三甲氧基硅烷反应后,表面的羟基数目明显减少。以上结果说明硅烷偶联试剂成功固载在GO表面,且烷基氯与三级胺原位生成了季铵盐。此外,GO-H-Bu4的季铵N处的峰明显高于GO-H-Bu1,说明利用正溴丁烷实现了进一步的季铵化。图中GOH-Bu1和GO-H-Bu4的Si-OH特征峰是由于部分硅烷水解产生[12-13]。2 400~2 300 cm-1处特征峰为CO2不对称伸缩振动,这是由于GO、GO-H-Bu1和GO-HBu4吸附空气中CO2导致,说明GO及其功能化材料能较好的吸附CO2[12-13]。
图3是GO和GO-H-Bu4的XPS全谱图,其中C与O的比值(n/n)分别为2.1和3.4。说明功能化后GO表面的含氧基团明显减少,这是由于利用GO表面的羟基引入了硅烷偶联试剂和Hatm[12]。GO-HBu4中除了含有 O和C外,还含有 Si、N和I,进一步说明成功地将季铵盐嫁接到GO表面。
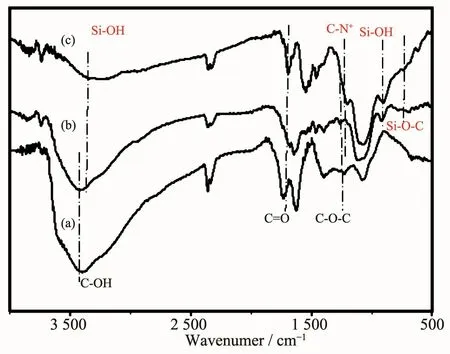
图2 (a)、(b)和(c)分别为 GO、GO-H-Bu1和 GO-H-Bu4的FT-IR谱图Fig.2 FT-IR spectra of(a)GO,(b)GO-H-Bu1and(c)GO-H-Bu4
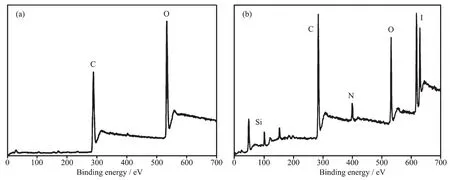
图3 (a)GO和(b)GO-H-Bu4的XPS全谱图Fig.3 XPS survey spectra of(a)GO and(b)GO-H-Bu4
进一步对GO-H和GO-H-Bu4的N1s谱图进行分峰拟合,探究材料表面的胺物种及其含量(图4)。N1s谱图分峰拟合结果显示,材料表面存在2类不同的氮物种,其中399.6 eV特征峰处归属于三级胺[12-13];401.1 eV处特征峰归属于季铵N,说明烷基氯和Hatm中的三级胺反应成功地将季铵盐引入到GO表面,GO-H和GO-H-Bu4中季铵N含量分别为N物种的24.8%和47.3%。由此计算可知,GO-H和GO-H-Bu4表面Hatm分子中被季铵化的三级胺的平均数目分别为0.99和1.89(表1)。
表1是 GO、GO-H、GO-H-Bu4的 XPS和元素分析结果。N原子在GO-H和GO-H-Bu4中(不考虑H)质量分数约为7.77%和7.19%。由此计算可知,GOH和GO-H-Bu4中Hatm负载量约为1.39和1.18 mmol·g-1,其季铵盐的负载量等于Hatm负载量与Hatm分子中被季铵化的三级胺的平均数目的乘积,分别为1.38和2.23 mmol·g-1;其三级胺的负载量等于4倍的Hatm负载量减去季铵盐的负载量,分别为4.18和2.49 mmol·g-1。该结果表明,利用多阳离子化的策略使得GO表面ILs的固载量显著提高,且构建的F-GO表面富含季铵盐和三级胺。
采用BET法分析了GO-H-Bu4的比表面积和孔结构,其结果如图5所示。由图可知,p/p0在接近0时,增加压力,吸附量增加,等温线略微上凸;但p/p0达到0.1后,继续增加压力至p/p0为0.4时,吸附量变化并不明显,说明较低压力下以单层吸附为主;当相对压力至0.8后,继续增加压力,吸附量显著增加,且在相对压力接近1时急剧升高,且出现毛细凝结导致的滞后环,这是典型的Ⅳ型吸附等温线。此外,GO-H-Bu4比表面积为16 m2·g-1,其内部微孔面积为 3 m2·g-1,外表面积为 13 m2·g-1。
图6为GO-H-Bu4的热重曲线。由图可知,GOH-Bu4在182℃之前失重约为5.7%(w/w)。由FT-IR谱图可知,2 400~2 300 cm-1处出现CO2不对称伸缩特征峰,3 380 cm-1附近出现自由水的特征峰。由此说明,上述失重是由吸附在GO-H-Bu4表面的CO2和H2O在受热后脱附引起。在182℃之后出现明显的热失重,可能为GO表面含氧基团、Hatm骨架以及硅烷的热分解导致。以上结果说明GO-H-Bu4有一定的热稳定性。
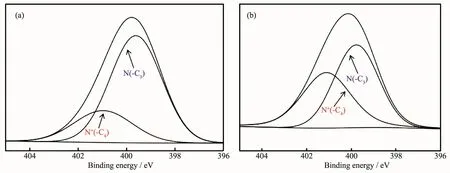
图4 (a)GO-H和(b)GO-H-Bu4的N1s分峰拟合后的高分辨图谱Fig.4 N1s high resolution spectra of(a)GO-H and(b)GO-H-Bu4

表1 GO和功能化GO的XPS和元素分析结果Table 1 XPS and elemental analysis results of GO and functionalized GO

图5 GO-H-Bu4的N2吸附-脱附等温线Fig.5 N2adsorption-desorption isotherm of GO-H-Bu4
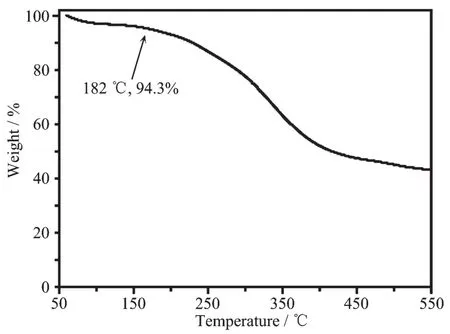
图6 GO-H-Bu4的热重曲线Fig.6 TG curve of GO-H-Bu4
2.2 催化剂的活性评价
以CO2和PO环加成反应为模型反应比较多元催化体系的催化性能(表2)。由表可知,分别以Hatm与 GO、GO与 n-BuBr、Hatm 与 n-BuBr组成双组份催化剂,反应后得到少量的碳酸丙烯酯(PC);以GO、Hatm与n-BuBr三组分催化时,活性显著提高,PC收率增加至55.0%,这可能是由于Hatm与n-BuBr原位形成季铵盐,进而与GO表面的羟基协同作用促进了环加成反应;随着n-BuBr用量的增加,催化活性显著提高,说明卤素阴离子对环加成反应至关重要。
随后,考察了季铵盐功能化GO多相催化剂的催化性能。由表3可知,单独的GO对环加成反应几乎没有活性,以Hatm为前驱体制得的GO-H为催化剂时,PC收率达82.9%。比较不同三级胺为前驱体制得的功能化GO的催化活性,发现催化活性依次为 GO-H>GO-D>GO-T>GO-TEA。以三乙胺为前驱体合成的GO-TEA活性较低,可能是因为其表面几乎没有碱性位。Hatm富含叔胺且具有笼状结构,有利于CO2的吸附,其被固载后单分子中仍有3个叔胺,其三级胺含量高达4.18 mmol·g-1,故催化活性最高。实验结果说明碱性位的存在有利于环加成反应进行。GO、Hatm与n-BuBr三元催化剂本质是Hatm和n-BuBr原位生生成的季铵盐与GO共同催化环加成反应,该催化体系催化活性明显低于GOH和GO-H-Bu4催化剂。这可能是GO-H和GO-HBu4表面引入硅羟基促进了环加成反应。此外,在催化剂洗涤过程中加水洗涤的主要目的是让部分未反应的Si-OCH3水解,引入了更多的硅羟基,其制得的催化剂催化活性明显高于未加入水洗涤制备的催化剂(表3,Entry 2和3)。由此说明硅羟基对促进CO2环加成反应也至关重要。
为了证明“多阳离子化”的策略有利于提高CO2环加成催化剂的催化活性,以n-BuBr对功能化的GO进一步季铵化,其催化剂催化活性如表4所示。由表可知,以四甲基乙二胺、三乙烯二胺和Hatm为前驱体,加入正溴丁烷进一步季铵化后,活性均有所增加,说明“多阳离子化的策略”有利于提高CO2环加成反应催化活性;随着n-BuBr加入量的增加,催化剂活性逐渐提高,当n-BuBr与三级胺前驱体物质的量比值为1时,活性增加不明显;当n-BuBr用量增加至三级胺前驱体物质的量的4倍时,催化剂催化活性显著增加。元素分析和XPS结果显示,n-BuBr加入量为8 mmol时制得的催化剂GO-H-B4表面季铵盐固载量达2.23 mmol·g-1,明显高于GOH(1.38 mmol·g-1)。尽管GO-H表面的三级胺固载量(4.18 mmol·g-1)明显高于GO-H-B4表面的三级胺固载量 (2.49 mmol·g-1),但GO-H-B4的活性明显高于GO-H。季铵盐在CO2环加成反应中主要作用为促进环氧化物开环活化,由此说明环氧化物的开环活化为CO2环加成反应的速率控制步骤[15-17]。FT-IR、XPS和元素分析结果显示GO-H-B4表面富含硅羟基、三级胺和季铵盐。结合文献报道[15-17]与实验结果说明依据氢键给体、卤素阴离子和胺协同催化CO2环加成反应的设计思想可设计高效的环加成催化剂。
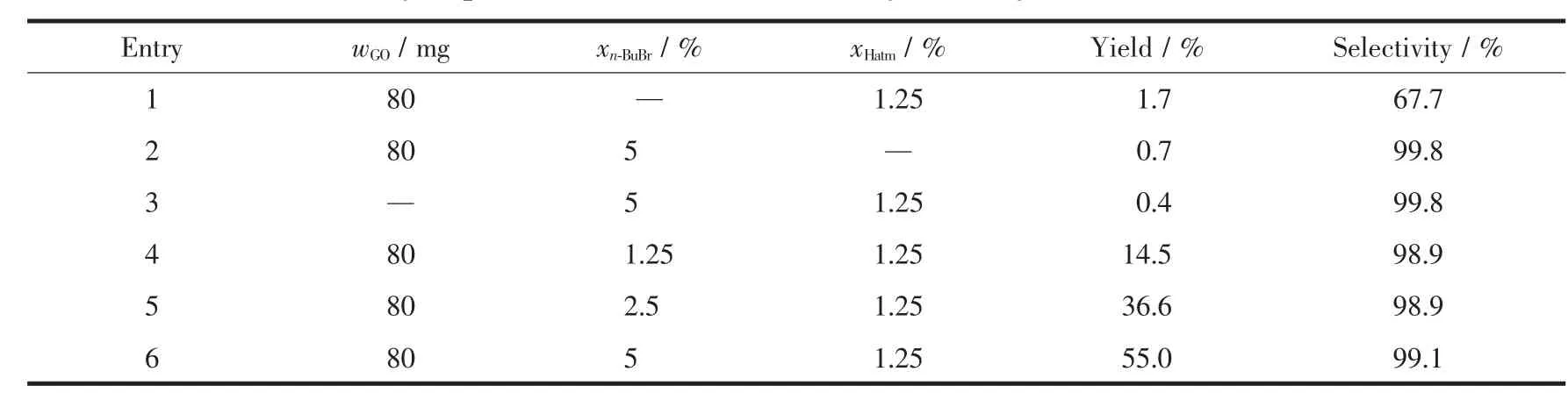
表2 比较不同催化体系对CO2环加成反应的影响Table 2 Catalytic performance of different catalysts for cycloaddition of CO2and PO

表3 不同碳材料催化剂催化CO2和PO反应的催化活性Table 3 Comparison of different carbon catalysts for cycloaddition of CO2to PO
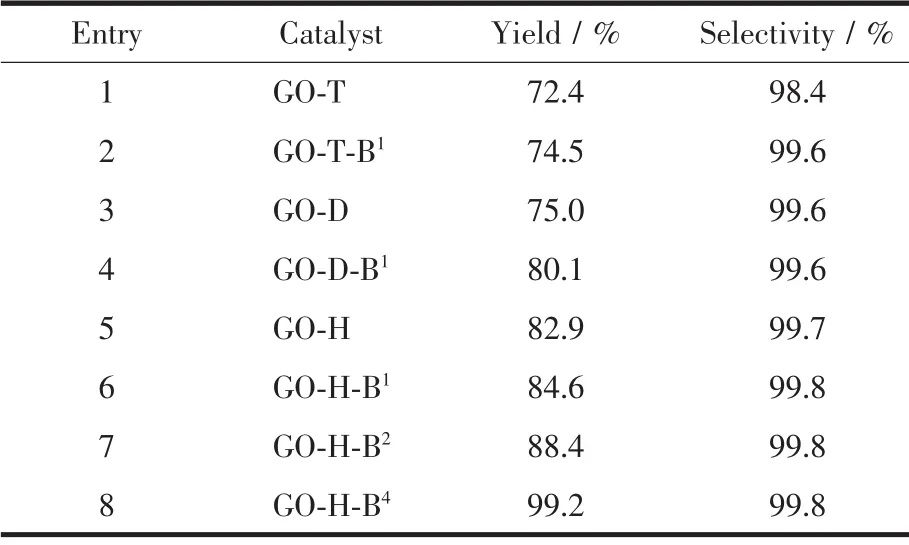
表4 不同功能化GO的催化活性Table 4 Catalytic performance of functionalized GO
适量的水对环加成反应有促进作用[18-20]。为了研究GO-H-B4耐水性,考察了水的加入量对环加成反应的影响。图7a表明PC收率先随着H2O加入量增加而提高,这可能是少量的H2O有利于GO-H-B4表面形成更多的C-OH和Si-OH,而C-OH和Si-OH作为氢键给体可与PO中O形成氢键降低PO开环的活化能,有利于促进CO2环加成反应;当H2O添加量达到10%(n/n)后,继续增加至15%(n/n)时,PC收率和选择性略有下降;但H2O添加量达到15%(n/n)后,继续增加其用量,PC收率和选择性急剧下降,这可能是由于水增加至一定浓度后,PO水解反应加剧。考虑到工业上PO中H2O含量远小于10%(n/n),所以GO-H-B4具有较好的耐水性。
由图7b可知GO-H-B4循环使用6次后,活性无明显下降,说明催化剂具有较好的循环使用性能。另外,从TG图可知,GO-H-B4在182℃后才发生热分解,而反应温度远低于182℃,由此说明该催化剂具有较好的稳定性。
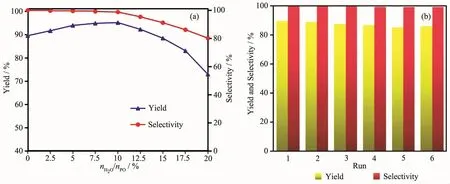
图7 (a)水添加量对环加成反应的影响;(b)GO-H-B4的循环使用性能Fig.7 (a)Effect of H2O addition on the catalytic performance of GO-H-B4;(b)Reusability test of GO-H-B4
2.3 可能的反应机理
Yin等[11]报道氢键给体与环氧化物中O形成氢键降低活化能,进而促进环氧化物在卤素阴离子作用下开环活化。胺可吸附并活化CO2,有利于CO2在环氧化物开环中间体中的插入反应。Wang等[21]报道胺不仅可与卤素阴离子协同作用,还能与氢键给体共同促进CO2环加成反应。由FT-IR和XPS等表征证明GO-H和GO-H-B4表面富含硅羟基、季铵盐和三级胺。通过对比实验证明硅羟基、季铵盐和三级胺对CO2环加成反应都至关重要。Liu等[22]证明分子内多氢键协同作用比分子间的氢键协同作用更有利于环加成反应。结合实验和表征结果提出了硅羟基、卤素阴离子和三级胺三者协同的催化机理如图8所示。首先PO与硅羟基形成氢键从而被活化,同时CO2被具有笼状结构的六次甲基四胺中的叔胺吸附并活化;随后卤素阴离子亲核进攻PO位阻较小的亚甲基碳使PO开环形成烷氧基负离子中间体;接着被三级胺吸附活化的CO2插入到烷氧基负离子中间体形成碳酸鎓盐;最后,碳酸鎓盐分子内环化形成PC,催化剂GO-H-B4被释放。
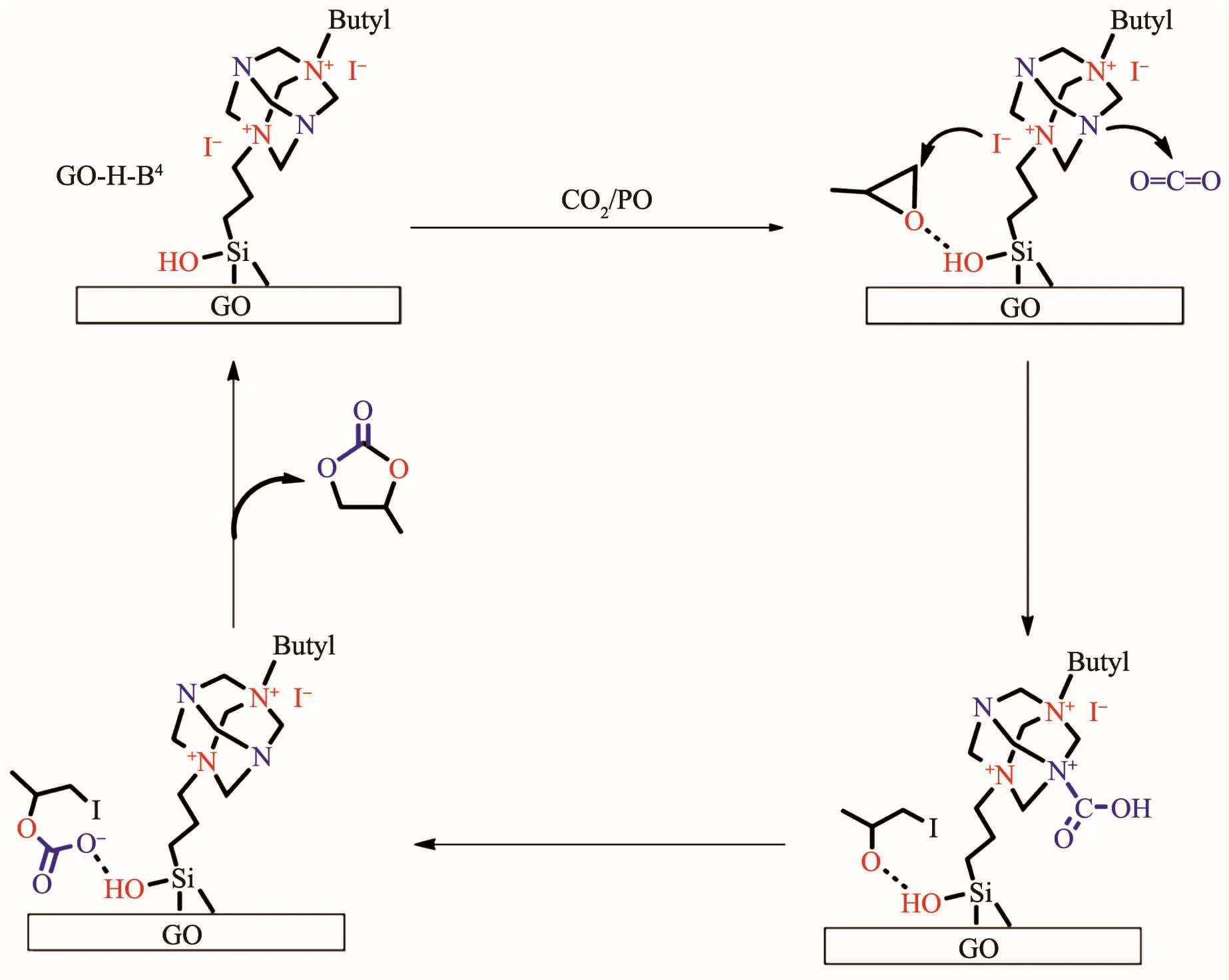
图8 GO-H-B4催化CO2环加成反应可能的催化机理Fig.8 Possible reaction mechanism for CO2cycloaddition over GO-H-B4
3 结 论
基于“多阳离子化”的策略,依据氢键给体、卤素阴离子和碱性位点协同催化的设计思想,设计并制备了硅羟基、三级胺和多阳离子季铵盐功能化GO多相催化剂,发现其在相对温和条件下可高效催化CO2和环氧丙烷环加成反应合成碳酸丙烯酯,且该催化剂具有较好的稳定性和耐水性。碱性位和季铵盐固载量对催化剂活性有较大影响,碱性位和季铵盐固载量越高,催化剂活性越高。“多阳离子化”的策略和“多基团协同作用”的思想可为构建高效的CO2环加成催化剂提供参考。
猜你喜欢
节能与环保(2022年3期)2022-04-26
波谱学杂志(2021年3期)2021-09-07
石油研究(2020年2期)2020-10-19
石油研究(2020年3期)2020-07-10
分析化学(2018年4期)2018-11-02
祖国(2018年17期)2018-10-30
艺术与设计·理论(2016年11期)2017-01-13
中学化学(2015年12期)2016-01-19
中学化学(2015年12期)2016-01-19
建筑工程技术与设计(2015年29期)2015-10-21
