
急性肾损伤肾小管上皮细胞修复的分子机制
2019-03-06 07:56:12综述李世军审校
肾脏病与透析肾移植杂志 2019年1期
姜 雪 综述 李世军 审校
急性肾损伤(AKI)最常见原因是缺血、中毒和梗阻等因素导致近端小管上皮细胞凋亡或坏死,其主要组织学特征包括近端小管上皮细胞刷状缘脱落,肾小管上皮细胞扁平和局灶性脱落,炎性细胞浸润以及形成富含Tamm-Horsfall蛋白的管型[1]。轻度损伤时,近端小管上皮细胞再生使肾小管形态和功能恢复。而在重度损伤区域,上皮细胞广泛受损、管周毛细血管损伤、内皮细胞及周细胞破坏、炎性细胞浸润、成纤维细胞和α平滑肌肌动蛋白(α-SMA)肌成纤维细胞增生,致使受损细胞外基质重塑不良、修复失调,造成持续的慢性炎症和纤维化[2]。肾小管萎缩、肾单位进一步丢失和纤维化的恶性循环,导致AKI逐步进展为慢性肾脏病。本文对AKI肾小管上皮细胞修复的分子机制进行综述,以期寻找促进近端小管再生的新治疗靶点,抑制其慢性炎症反应和纤维化进展。
肾小管上皮细胞与肾单位修复
小鼠AKI模型的遗传命运图谱研究显示,残存的肾小管上皮细胞(tubular epithelial cells,TECs)增生修复损伤的肾小管,肾小管外的细胞群不参与上皮细胞修复,这项研究结果同样适用于人类[3]。但是,仍然存在由肾小管祖细胞介导上皮修复的可能性。究竟是残存的TECs还是肾小管祖细胞介导上皮修复仍有争论[4]。参与人和小鼠近端小管修复的CD133+CD24+细胞的发现增加了固有肾小管祖细胞群存在的可能性[5]。体外研究发现这些细胞具有极强的自我更新能力,并且可以经诱导产生多个克隆细胞系,但体内研究尚未完全证实。最近一项研究表明,与体外研究结果相比,体内大多数器官的周细胞不具有间充质干细胞样特性[6]。遗传谱系示踪技术是追溯不同细胞群转归的最可靠方法,但这项技术不能用于人体研究。Corbeil等[7]发现健康小鼠肾脏近端小管中CD133表达丰富,因此质疑这种细胞群的干/祖细胞标记是否准确。由于无法对人类体内CD133+CD24+细胞进行遗传命运图谱分析,且其在小鼠肾脏中的表达存在争议又缺乏准确的分子特征,故CD133+CD24+细胞在肾脏损伤修复中的机制研究仍有困难[4]。
研究发现损伤的上皮细胞中角蛋白19,Bcl2和波形蛋白表达上调[8]。在人类肾脏中,CD133+CD24+细胞表达波形蛋白,与CD133-CD24-的细胞相比,其细胞质、线粒体较少,刷状缘模糊。这些细胞主要于AKI后测出并表达肾损伤分子-1(Kim-1+)。Kusaba等[9]研究发现缺血再灌注损伤后小鼠CD24、CD133、Kim-1和波形蛋白表达上调。总之,这些研究结果表明CD133+CD24+细胞可能代表受损的TECs。持续多西环素标记实验显示,损伤前标记的TECs数量没有增加; 而损伤后标记细胞显著增加[10]。虽然这种标记无法准确证实幸存TECs群体的真正增殖方式,但表明很可能是由幸存受损上皮细胞进行AKI后的肾小管修复,一部分此类细胞表达Kim-1+Ki67+。
TECs在上皮内稳态和损伤后都严格保持其管状特征。R26VT2 / GK3小鼠研究显示在上皮内稳态和修复期间,克隆维系单个肾单位谱系和小管类型转归[11]。在上皮细胞稳态期间,Wnt反应细胞(Axin2+细胞)在肾皮质和髓质中大量复制,与ActinCre+细胞相比,它有更强的增殖能力。在横纹肌溶解AKI模型中,Axin2+细胞在损伤后2个月内表现出更大规模的克隆扩增。但AKI后Wnt反应细胞子代的细胞类型和Wnt配体的来源尚不清楚。这种细胞的遗传稳定性及其对肾脏修复的影响尚未验证,故而这些细胞在损伤后上皮再生中的作用机制仍不明确。损伤后对近端小管上皮细胞中Wnt4诱导提示Wnt通路与肾脏修复之间可能存在联系[12]。然而Wnt4的检测基于免疫染色,迄今为止,尚无检测Wnt蛋白的可靠抗体。随后的研究也未证实肾小管上皮细胞中Wnt4表达。相反,在缺血再灌注损伤后,Wnt4在髓质肌成纤维细胞中被重新激活,且去除Wnt4对纤维化或肌成纤维细胞增殖没有影响[13]。
目前,尚无确凿证据证实在肾单位中存在干细胞或祖细胞群。然而,仍然存在具有强大体内复制能力的这种特殊TECs的可能性。因此未来可期通过药物研发来激发具有潜在高复制能力的细胞类型以增强内源性修复过程。
促进肾小管上皮细胞修复的细胞因子
肾小管上皮细胞中是否存在独特的细胞类型仍存争议,AKI后上皮再生的关键细胞因子仍需进一步研究。损伤诱导Sox9激活是一种关键再生反应[14],研究显示Sox9是缺血再灌注损伤后24h内上调最高的转录因子之一[15]。正常肾脏的近端小管中仅有很少的Sox9+细胞,而在calbindin-28dk+的远曲小管上皮细胞中Sox9+细胞成簇存在[14]。在缺血和梗阻性AKI中,Sox9+细胞可用于区分受损的增殖性近端小管上皮细胞亚型,损伤后48h约40%至45%的Sox9+细胞与Ki67共表达。在AKI向慢性肾脏病转化模型中,研究人员证实Sox9是受损肾单位上皮细胞再生的关键内在活化因子[14]。损伤后早期激活Sox9细胞的命运图谱研究显示,其子代恢复了与正常肾小管上皮细胞相似的顶端-基底端极性,从而证实这些细胞可再生近端小管上皮细胞[14](图1)。与对照组小鼠相比,在缺血再灌注损伤后4周,近端小管特异性敲除Sox9的小鼠早期肾脏修复反应和肾功能恢复均严重受损,并产生更严重的纤维化。缺血再灌注损伤后4周,大部分近端小管中Sox9表达恢复到基线水平,近端小管恢复正常,损伤表达完全消除(Kim-1-)。然而,近端小管某些区域持续表达Kim-1,且这种Kim-1+的近端小管上皮细胞同时表达Sox9,此类Kim-1+Sox9+的近端小管上皮细胞亚型损伤修复尚未完成(图1)。此种细胞类型可能在AKI向慢性肾脏病转化中起作用[14]。Sox9也在中毒性AKI肾小管上皮细胞修复中发挥作用。Kang等[16]证明Sox9是中毒性AKI肾脏修复的关键细胞因子,但AKI后激活的Sox9+细胞或固有Sox9+细胞对整体修复的具体作用仍需进一步研究。
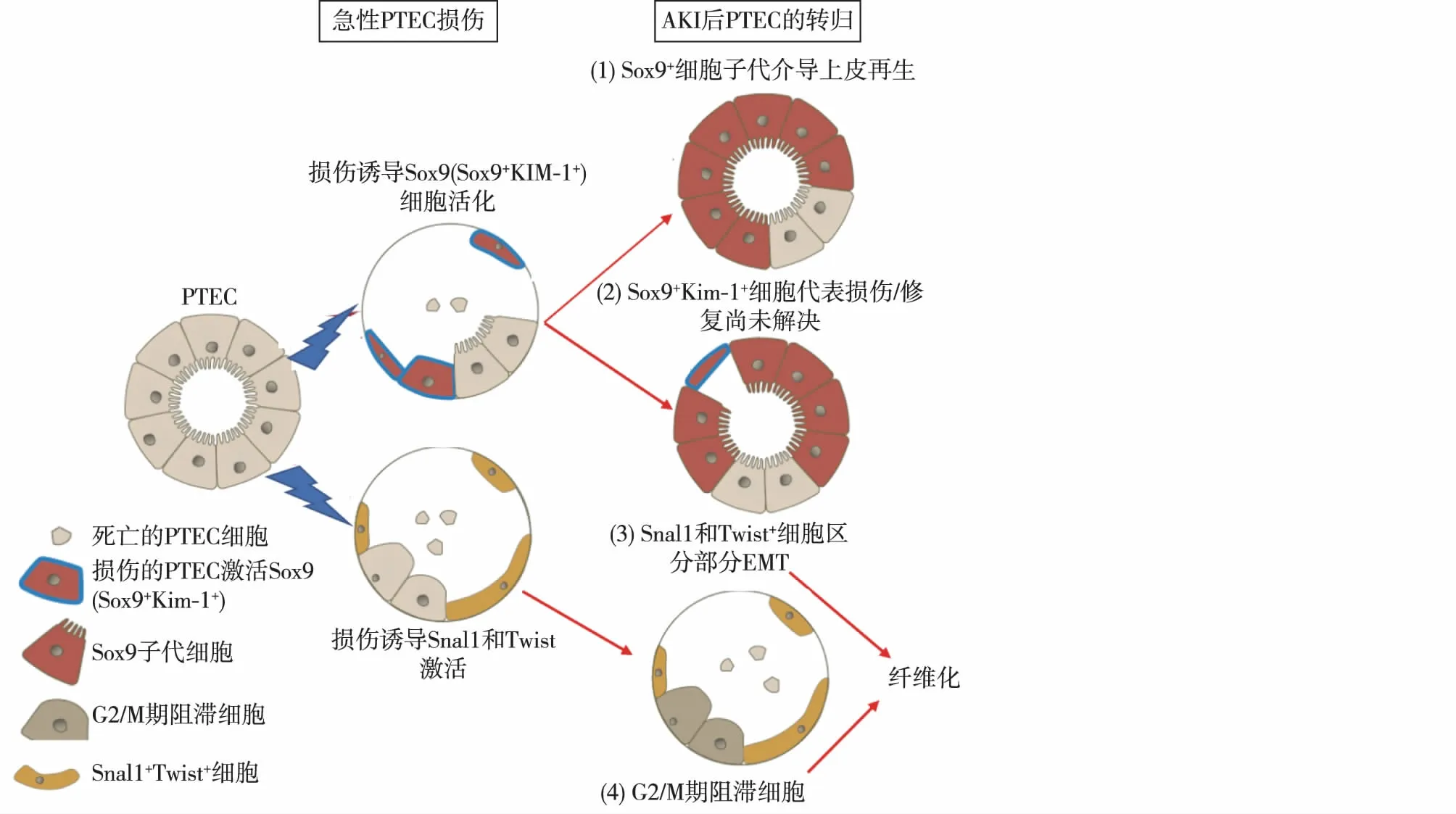
图1 AKI后PTEC的转归[21]PTEC:近端小管上皮细胞;AKI:急性肾损伤;KIM-1:肾损伤分子1;EMT:上皮-间充质转化
Sox9在肾小管上皮细胞形成和再生中均起着重要作用,在人类和小鼠肾脏胚胎发育中十分关键。单个Sox9等位基因失活突变可因其单倍体不足导致胃肠道发育不良综合征[17],其肾脏缺陷包括肾积水和肾发育不全。在肾脏胚胎发育过程中,敲除Sox8和Sox9可导致小鼠肾脏发育不全[18]。为了确定Sox9在近端小管上皮细胞修复和小管发育中的作用,研究人员对胚胎期肾脏Sox9+细胞进行遗传谱系示踪,结果显示Sox9+细胞可发育成大部分肾小管上皮细胞[14]。然而,重新激活的基因是否会通过与它在发育过程中相似的基因调控网络来再生受损的上皮细胞尚不明确[19]。与胚胎期相比,器官发生过程中起关键作用的活化基因于再生过程中可能会激活不同的基因调控网络,Sox9的作用机制中或许存在此种因素。去分化是将分化成熟的细胞恢复到其发育期间在该谱系中的原始形式。研究发现叶酸诱导AKI后重新表达Pax2,故而Pax2被视为“肾小管上皮细胞去分化标记物”[20]。正常成人肾脏中髓质集合管持续表达Pax2,损伤发生后,Pax2在近端小管上皮细胞中重新表达。AKI后,Pax2+细胞的转归及Pax2在上皮修复中的作用仍有待研究证实。同时Pax2或Sox9能否真正界定去分化的近端小管上皮细胞亚型尚不清楚。
急性肾损伤后,部分近端小管上皮细胞凋亡坏死,部分幸存近端小管上皮细胞产生多种损伤修复反应。(1)早期损伤修复反应:损伤诱导活化Sox9+Kim-1+的近端小管上皮细胞亚型,再生受损的上皮细胞[14]。(2)慢性损伤修复反应(小鼠缺血再灌注损伤后28d):成功再生的上皮细胞Sox9转阴; 而由Kim-1+的近端小管上皮细胞损伤尚未修复区域持续Sox9+应答试图自我再生。在此阶段,大多数增殖的Ki67+近端小管上皮细胞表达Sox9+; 因此,在慢性期,Sox9+Kim-1+细胞可区分尚未完全修复的近端小管上皮细胞亚型[14]。(3)损伤激活Snai1和Twist1使部分上皮细胞转化至间充质状态[22-23]。部分上皮-间充质转化(EMT)与病理性促炎和促纤维化反应有关。此类细胞的细胞周期阻滞。(4)小鼠重度AKI模型(单侧缺血再灌注损伤、双侧严重缺血再灌注损伤、单侧输尿管梗阻模型)和经典纤维化模型(马兜铃酸肾病)中,G2/M期阻滞的上皮细胞加剧了纤维化进展[24](图1)。
阻碍肾小管上皮细胞修复的细胞因子
虽然受损的TECs在细胞类型方面高度保真; 仍有少数细胞可发生EMT,影响肾小管上皮细胞修复,加重促炎和促纤维化反应[22-23]。从部分EMT状态治疗受损细胞可提高损伤后肾脏修复效率。损伤时,TECs随机发生部分EMT,同时分泌促炎和促纤维化因子。已有研究表明损伤激活的Snai1是胚胎发育和肿瘤进展期间有效的EMT诱导剂,可诱导部分EMT。单侧输尿管梗阻和叶酸诱导的AKI中,与对照组相比,近端小管上皮细胞特异性Snai1基因敲除动物的炎症反应、α-SMA+肌成纤维细胞数量及纤维化程度明显减弱[23]。另一项研究表明,在缺乏Snai1和Twist1(一种关键的EMT调节因子)的小鼠中,AKI所致的纤维化程度显著降低[23]。这项研究显示,Snai1和Twist1可导致TECs的G2期阻滞,从而抑制TECs的增殖并增强其促炎和促纤维化反应。部分EMT可能是严重受损的TECs尝试获得迁移表型以覆盖裸露肾小管基膜的表现。然而,在这种损伤修复反应过程中,同时启动了促炎和促纤维化分子程序。与完全EMT相比,发生部分EMT的受损TECs除具有迁移特性外,还获得了转分化为间质肌成纤维细胞的侵袭性表型[25]。Yang等[24]发现G2 / M细胞周期阻滞的近端小管上皮细胞可以分泌促炎和促纤维化细胞因子,加重纤维化程度。总的来说,这些研究主张更全面地了解影响肾小管上皮细胞修复的细胞因子,以期寻找干预其促炎促纤维化过程的靶向药物,减少AKI进展为慢性肾脏病。
调节肾脏修复的细胞分子信号通路
AKI后肾脏修复可能是损伤诱导的多种信号通路激活、关键下游分子驱动和/或阻碍受损上皮细胞再生之间相互作用的结果。损伤诱导表皮生长因子受体(epidermal growth factor receptor ,EGFR)、集落刺激因子1(colony stimulating factor 1,Csf-1)、经典Wnt/β-catenin通路和视黄酸信号传导通路(RA signaling)基因扰动,影响AKI后肾脏修复。研究表明EGFR和Erbb2是缺血性AKI后小鼠肾皮质磷酸化最多的磷酸受体酪氨酸激酶[26]。抑制表皮生长因子受体磷酸化或近端小管上皮细胞特异性敲除表皮生长因子受体,均可导致近端小管上皮细胞增殖减少、早期功能和组织学修复受损。虽然许多研究表明缺血、中毒和梗阻性AKI模型中通过配体(如外源性表皮生长因子或肝素结合表皮生长因子)激活表皮生长因子受体通路具有修复作用[27],但其修复过程中的下游机制仍有待阐明。近端小管上皮细胞中损伤诱导的STAT3-Birc5信号传导通路似乎亦是一种修复反应。γ-分泌酶抑制剂抑制STAT3(信号传导及转录激活因子3)磷酸化可导致Birc5表达下调、修复反应延迟[28]。与对照组相比,近端小管上皮细胞特异性Birc5敲除小鼠的TECs增殖减弱、肾功能和组织学恢复延迟。
AKI后,TECs中表皮生长因子受体和STAT3/ Birc5信号传导被激活。肝素结合表皮生长因子 (HBEGF)是上皮细胞释放的表皮生长因子受体信号传导配体,以自分泌或近分泌方式激活表皮生长因子受体信号传导通路。巨噬细胞通过旁分泌促进上皮细胞中的集落刺激因子1(Csf-1)激活,Csf-1的激活反过来促进M2巨噬细胞表型标志物生成。巨噬细胞旁分泌激活TECs中的RA信号传导通路和经典Wnt/β-catenin信号通路。巨噬细胞和内皮细胞分泌的IL-22和MMP(基质金属蛋白酶)分别作用于TECs促进肾脏修复。内皮细胞通过S1pr1(鞘氨醇1-磷酸酯受体1)和Hif1α/ Hif2α(缺氧诱导因子)信号传导调节慢性炎症反应,促进修复。此外,血管分泌因子如VEGF、Ang1和Sirt1(组织沉默调节蛋白1)均影响肾脏修复反应[21],但目前仍需进一步研究加以证实(图2)。

图2 肾脏修复的细胞分子信号网络[21]
小结:对AKI后肾小管上皮细胞修复的研究已有一定进展。损伤诱导肾小管上皮细胞再生、部分EMT和细胞周期阻滞体现了受损近端小管上皮细胞的多样化转归。目前对这些重要生物学过程的分子机制仍然知之甚少。对其机制的进一步研究可能会提示新的治疗策略和方向,从而发现肾脏修复过程中有希望的治疗靶点和生物标志物,以促进肾脏自身修复、抑制其伴随的纤维化,减少AKI进展为慢性肾脏病。
猜你喜欢
辅导员(2020年6期)2020-04-23 12:43:12
派出所工作(2018年4期)2018-09-10 06:40:58
飞碟探索(2016年11期)2016-11-14 19:33:44
中外医疗(2015年11期)2016-01-04 03:58:45
中国医药生物技术(2015年4期)2015-12-26 08:26:36
中国现代医学杂志(2015年26期)2015-12-23 11:04:22
医学研究杂志(2015年8期)2015-06-22 14:00:57
医学研究杂志(2015年12期)2015-06-10 06:57:46
西南国防医药(2015年11期)2015-02-28 19:38:46
西南军医(2015年6期)2015-01-23 01:25:49
