
高效液相色谱-串联质谱法同时测定葡萄酒中甜味剂、防腐剂和色素
2019-02-25 02:08:12王丽娟柯润辉安红梅杨春艳山树棠尹建军
酿酒科技 2019年1期
王丽娟 ,柯润辉 ,安红梅 ,杨春艳 ,山树棠 ,尹建军
(1.中国食品发酵工业研究院有限公司,北京100015; 2.国家食品质量监督检验中心,北京100015)
食品添加剂是现代食品工业中不可缺少的一部分,人工合成色素、甜味剂和防腐剂作为重要的食品添加剂被广泛应用于食品生产加工[1-2]。但是,随着其在食品行业的广泛使用,违规及超限量使用事件时有发生,所带来的食品安全问题也日益凸显,安全监管问题已被列入食品安全整顿工作的重要内容[3-4]。随着人们生活水平的提高及对健康的关注,葡萄酒慢慢取代了酒精度较高的白酒,成为了很多宴请和聚餐的“新宠”,消费量呈现日益增长的趋势[5],但葡萄酒市场仍有不少违法使用添加剂或者添加剂超标的现象,甚至有不法商贩利用香精、色素、酒精、甜味剂等添加剂进行勾兑,制造假冒伪劣葡萄酒产品,扰乱葡萄酒市场,危害消费者健康[6-7]。近年来,国家有关部门在对全国葡萄酒的抽检过程中发现,滥用添加剂现象时有发生,因此葡萄酒中违规使用的添加剂也成为国家食品安全监督抽检的重点项目。随着我国食品安全监管水平的提升,为提高工作效率,方便监管部门和生产企业对葡萄酒中防腐剂、甜味剂和色素的日常监测,开发灵敏度高、适用性广的高通量检测方法以有效地应对添加剂在葡萄酒中的滥用,以及应对突发食品安全事件具有重要的现实意义[8]。
为保障食品安全,需要对葡萄酒中食品添加剂的含量进行检测。目前,国标[9-11]有针对葡萄酒中防腐剂、色素和甜味剂等的检测方法,但这些标准大都针对单一或某几种食品添加剂,且样品的预处理方法各不相同,对于葡萄酒中多种添加剂则需要逐一测定,程序繁杂,检测时间长,效率很低[6,12],在进行批量样品检测时,需要大量的人力物力。近年来,国内外关于葡萄酒中防腐剂、甜味剂和色素检测方法的文献报道很多,主要采用离子色谱法[13]、气相色谱法[14]、高效液相色谱[15-16]、高效液相色谱-质谱法[17-18]等,虽然相关检测方法的报道很多,在检测灵敏度和准确度方面均有所提高,但经文献检索,大多只针对某一类添加剂进行检测,不利于对葡萄酒中多类添加剂的大规模筛查。另外,包括我国国家标准在内,目前最为普遍的分析方法为液相色谱法[19-20],该方法不能提供目标化合物的化学结构信息,抗干扰能力相对较弱,在实际样品分析时容易受到基质干扰而产生假阳性现象[5,21]。液相色谱与质谱联用技术能够在一定程度上降低基质干扰,对于需要高灵敏度、宽适用范围的检测工作而言,已成为最佳的手段之一,逐渐应用于添加剂的检测[22]。本实验采用高效液相色谱-串联质谱技术,利用高效液相色谱的强分离能力和质谱准确定性、高选择性和高灵敏度的特点,在较短的时间内即可完成多组分的分离鉴定,建立了一种可同时测定并能够准确定性定量葡萄酒中防腐剂、甜味剂和色素的方法。
1 材料与方法
1.1 材料、试剂与仪器
葡萄酒样:购自零售超市和商店。
仪器设备:LCMS-8050高效液相色谱-串联质谱仪(日本Shimadzu公司,具体配置为LC-20ADXR×2输液泵,DGU-20A3R在线脱气机,SIL-20AXR自动进样器,CTO-20AC柱温箱,CBM-20A系统控制器,LCMS-8050三重四极杆质谱仪,Lab-Solutions Ver.5.91色谱工作站);Milli-Q Reference超纯水器(美国Millipore公司);超声波清洗器(KQ-500DE,昆山超声仪器有限公司);Acquity UPLC HSS T3柱(100 mm×2.1 mm,1.8µm,美国Waters公司);0.22 μm尼龙滤膜(上海安谱科学仪器有限公司)。
试剂:甲醇、乙腈、乙酸、乙酸铵(HPLC级,Dikma公司);甜蜜素(Sodium cycamate)、安赛蜜(Acesulfame-K)、糖精钠(Saccharin sodium)、阿斯巴甜(Aspartame)、三氯蔗糖(Sucralose)、甜菊糖苷(Stevioside)、纽甜(Neotame)、阿力甜(Alitame);诱惑红(Allura red)、赤藓红(Erythrosine)、苋菜红(Amaranth)、胭脂红(Carmine)、柠檬黄(Tartrazine)、日落黄(Sunset yellow)、亮蓝(Brilliant blue);脱氢乙酸(Dehydroacetic acid),美国Sigma-Aldrich公司,加拿大TRC公司和德国Dr.Ehrenstorfer GmbH公司,纯度≥95%。
1.2 实验方法
1.2.1 标准溶液的配制
分别准确称取16种添加剂标准品于10 mL棕色容量瓶中,用水或甲醇溶解并定容,配制成质量浓度为1 mg/mL的单标储备液,于-18℃下避光保存。用甲醇稀释并配制中间浓度的标准工作液,于4℃避光保存。根据需要用流动相逐级稀释,配制成适当浓度的混合标准工作液,现配现用。
1.2.2 样品前处理方法
取葡萄酒样品2.0 mL于10 mL容量瓶中,用超纯水定容,超声混匀,过0.22 μm滤膜,滤液供测定。
1.2.3 色谱条件
色谱柱:Acquity UPLC HSS T3柱(100 mm×2.1 mm,1.8 µm,美国Waters公司)。流动相:(A)甲醇和(B)10 mmol/L乙酸铵水溶液,流速0.35 mL/min;梯度洗脱程序为:0 min,甲醇的体积分数为10%;0~3 min,甲醇的体积分数从10%升至90%,并保持2.5 min,5.5~5.51 min;甲醇的体积分数从90%降至10%;5.51 min~6.5 min,甲醇的体积分数保持10%。柱温为35℃;进样体积:2 μL。
1.2.4 质谱条件
分析仪器:LCMS-8050;离子源:电喷雾离子源(ESI);扫描方式:正负离子同时扫描;离子源接口电压:0.5 kV;雾化气:氮气3.0 L/min;干燥气:氮气10 L/min;碰撞气:氩气;DL温度:250℃;加热模块温度:400℃;扫描模式:多反应监测(MRM);驻留时间:15 ms;延迟时间:3 ms;16种添加剂标准品MRM参数见表1。
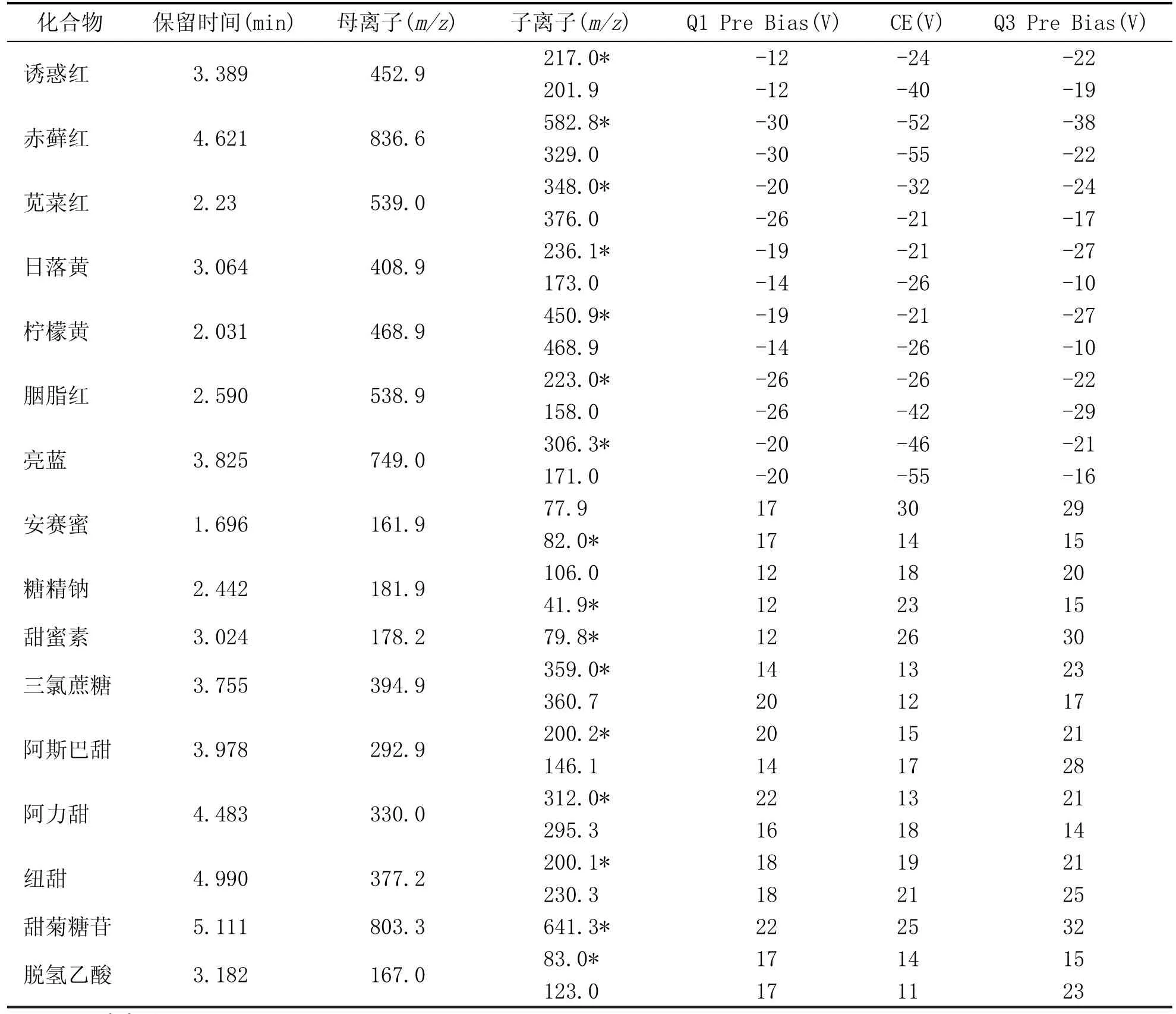
表1 16种添加剂的保留时间和质谱条件
2 结果与讨论
2.1 样品前处理条件的优化
与其他分析手段不同,质谱分析往往存在一定的基质效应,当使用ESI源时,在离子化过程中基质组分与目标物竞争,更易出现基质抑制效应。减少基质效应的方法通常有稀释样品溶液、增加净化步骤、配制基质匹配标准溶液等[23]。本研究采用直接进样方式进行分析,以避免传统的液液萃取、固相萃取方法回收率低、重复性差,且会使用大量溶剂的弊端,但考虑到葡萄酒中含有一定比例乙醇和糖类,为减少基质效应对测定的影响,需对样品进行稀释处理。稀释的方法可以降低乙醇对色谱峰形影响,减弱基质效应,但是会降低方法的检出限。经过对主流品牌的葡萄酒分别稀释2~15倍进行检测,不同稀释倍数样品测试对比,最终确定稀释倍数为5倍。
2.2 色谱和质谱条件的优化
2.2.1 流动相的选择
反相色谱的流动相通常由水和有机溶剂(如甲醇、乙腈)等组成,由于某些被测物极性较强,流动相的洗脱能力不宜过强,同时待分离的16种添加剂有些极性相近,保留时间相近,采用乙腈为有机相时导致几个组分“共流出”情况较甲醇严重,而甲醇可使16种添加剂的保留时间相对延迟,同时甲醇可以提高离子化程度,峰面积比用乙腈为流动相时大,故选择甲醇为实验的有机相;又由于流动相要进入质谱仪,添加一定量的缓冲溶液可以增加响应值,所以考虑甲醇-甲酸水溶液、甲醇-乙酸铵溶液、甲醇-乙酸水溶液3种流动相体系。以被测物在色谱柱上的分离度、峰形、灵敏度等为考察指标对3种流动相体系进行了比较。结果表明,体系中添加了乙酸铵不仅更有利于分离物质在色谱柱上的保留,而且能够提高色素类物质的电离化程度,增加信号响应,从各物质分离度、峰形、响应值以及保留时间的稳定性等指标进行综合衡量,甲醇-10 mmol/L乙酸铵作为流动相时,优于其余两种流动相,故实验选用甲醇-10 mmol/L乙酸铵为流动相进行梯度洗脱。
2.2.2 柱温的选择
柱温能够影响色谱柱的柱效、选择性、灵敏度和稳定性,柱温的改变直接影响分离效果及分离速率。升高柱温,色谱柱内离子交换速率随之升高,有利于提高柱效、缩短分析时间[24]。本实验考察了柱温分别为30℃、35℃、40℃时,16种添加剂保留时间及响应值的差别,发现当柱温为30℃时,16种添加剂有13种出峰时间均集中在3~4 min,分离度差;提高柱温至35℃时,柱效提高,分离度和灵敏度均有改善,继续提高柱温,分离情况无明显变化。考虑到色谱柱在相对较低温度下使用寿命较长,本实验设定柱温为35℃。优化条件下16种添加剂的MRM质谱图见图1。
2.2.3 质谱条件的选择
为获得最佳的灵敏度和分离效果,根据16种添加剂的分子结构特征,在正、负两种电离模式下优化被测物的母离子和特征子离子以及相应的质谱参数。取质量浓度为1 mg/L的16种化合物单标准溶液依次采用不接色谱柱直接进样方式进行质谱全扫描检测,得到目标分析物一级质谱图,再用氩气轰击该母离子,得到其二级质谱图,利用仪器的自动优化功能,分别对Q1、Q3、CE等进行优化,确定16种添加剂的母离子和子离子的最佳质谱条件,以强度较大的子离子作为定量离子,强度稍小的子离子为定性离子,各化合物的定量离子及定性离子见表1。在优化的色谱和质谱条件下16种被测物的总离子流(TIC)图见图1。
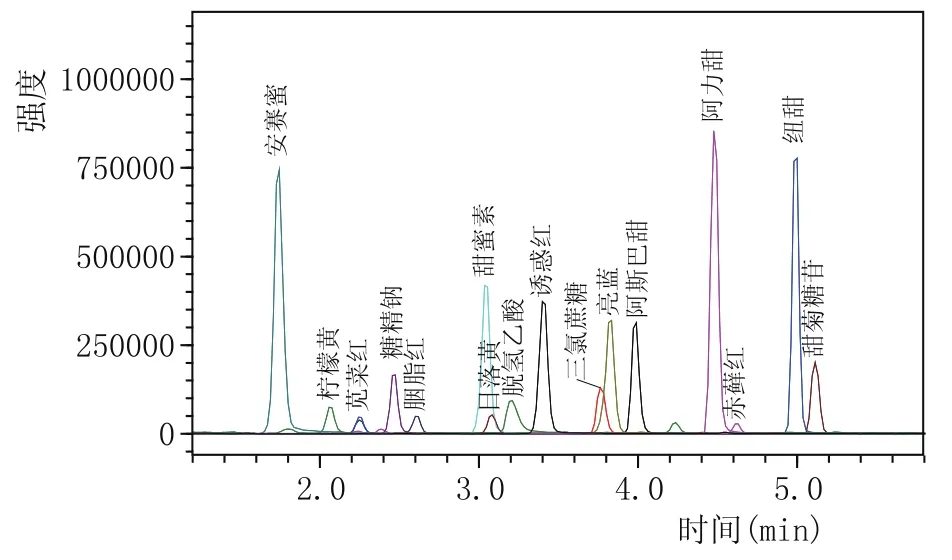
图1 16种添加剂混合标准溶液的总离子流图
2.3 方法学评价
2.3.1 方法的线性范围和检出限
将含16种被测物的混合标准储备液添加至稀释5倍的空白葡萄酒样品中,分别配制系列混合标准工作液,按1.2部分所确定的色谱和质谱条件进行检测,以各组分定量离子色谱峰面积对相应的质量浓度绘制各被测物的标准工作曲线,结果表明,线性关系良好,相关系数(r2)都在0.995以上。
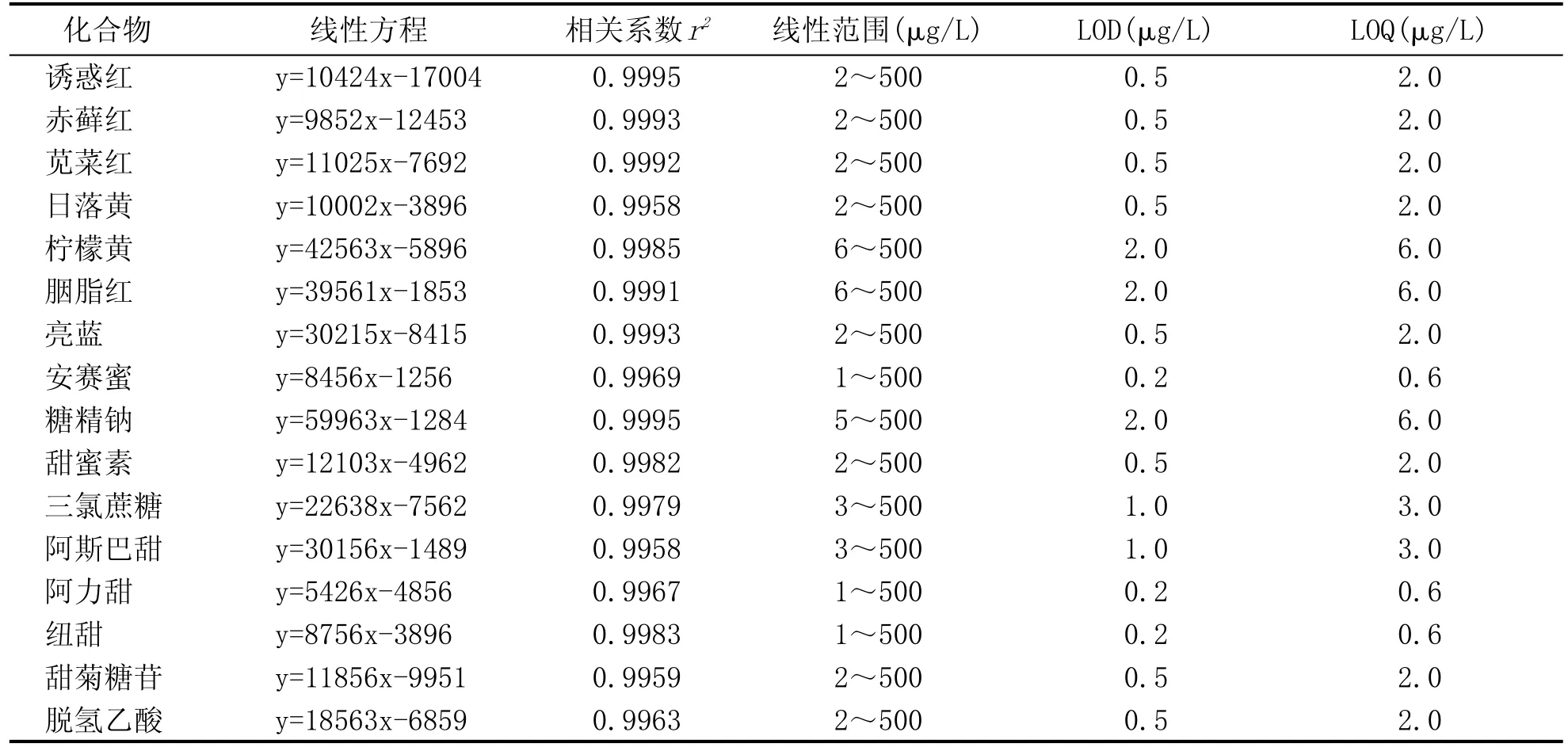
表2 16种被测物的线性范围、线性方程、相关系数、检出限和定量限
用葡萄酒样品低加标水平计算各组分的检出限和定量限,用信噪比为3确定方法的检出限(LOD),用信噪比为10确定方法的定量限(LOQ),各组分在葡萄酒样品中的检出限范围在0.2~2 μg/L之间。结果见表2。
2.3.2 精密度和回收率
用微量移液器向空白葡萄酒样品中准确加入一定量的16种待测物混合标准溶液,配成低、中、高3个浓度水平进行回收率实验,按1.2节所述实验步骤进行处理和测定,进行6次重复实验,计算其回收率及相对标准偏差(RSD)。结果表明,各目标物平均回收率在89.1%~106.4%之间,相对标准偏差小于9.7%,方法的准确度和精密度均符合多残留分析的要求。
2.4 实际样品的测定
按所建立的方法对采购的10批次葡萄酒样品进行了16种添加剂的筛查,每个样品重复测定3次。所检葡萄酒样品中有1个检出糖精钠和甜蜜素,含量分别为26.3 μg/L、38.9 μg/L,其余样品的检测结果均为阴性。阳性样品的MRM质谱图见图2。
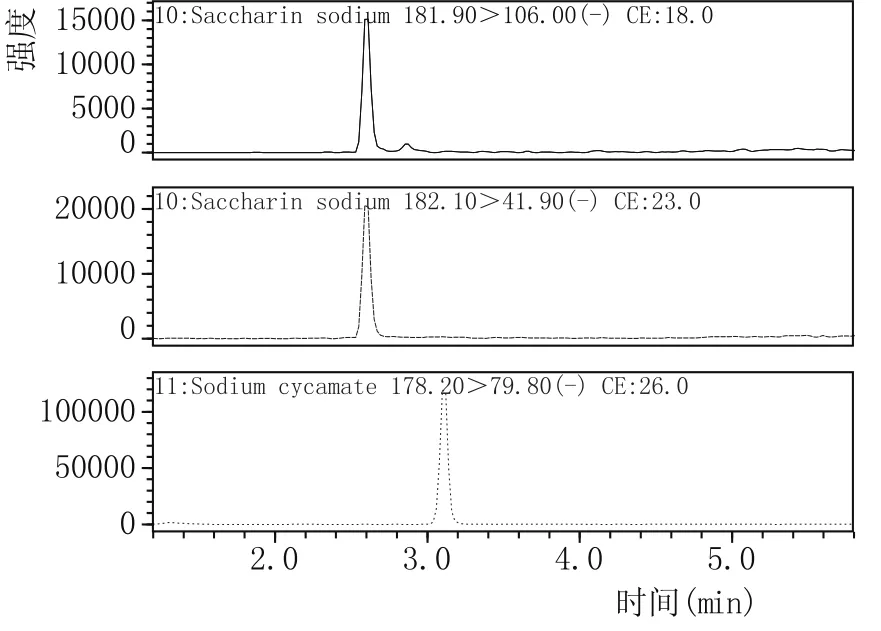
图2 某葡萄酒样品中检出的糖精钠和甜蜜素质谱图
3 结论
本研究建立了高效液相色谱-串联质谱仪同时测定葡萄酒中甜味剂、防腐剂和色素含量的方法,前处理简单、分析速度快、准确度和灵敏度高。实际样品的检测表明,该方法能够满足葡萄酒甜味剂、防腐剂和色素残留的分析要求,与国标方法相比,大大提高了分析效率和降低了分析成本,能够满足葡萄酒中痕量分析要求,非常适合大批量葡萄酒样品中16种添加剂的定性和定量分析。
猜你喜欢
基层中医药(2021年3期)2021-11-22 08:08:04
广州化工(2020年20期)2020-11-02 03:02:52
收藏界(2018年3期)2018-10-10 05:34:08
中国商界(2017年4期)2017-05-17 04:36:48
少年科学(2015年10期)2015-10-31 04:19:47
分析化学(2015年6期)2015-06-18 10:28:17
江苏调味副食品(2015年1期)2015-02-28 01:56:34
中国质量与标准导报(2014年6期)2014-02-28 22:24:10
河南科技(2014年16期)2014-02-27 14:13:08
首都食品与医药(2013年24期)2013-10-19 11:56:34
