
Castleman disease presenting with jaundice: A case report and review of literature
2019-02-22 07:33:48BoZhaiHaiYangRenWeiDongLiShivaReddyShuJunZhangXueYingSun
World Journal of Clinical Cases 2019年3期
Bo Zhai, Hai-Yang Ren, Wei-Dong Li, Shiva Reddy, Shu-Jun Zhang, Xue-Ying Sun
Abstract BACKGROUND Castleman disease (CD) is a rare lymphoproliferative disorder that presents with various symptoms. CD accompanied with jaundice is uncommon since there are only 11 cases reported in the literature.CASE SUMMARY Here we report a 62-year-old woman who was admitted to the hospital with signs and symptoms of intermittent jaundice. Biochemical tests showed higher serum levels of total and direct bilirubin, and normal serum levels of tumor markers and interleukin-6. Contrast-enhanced computed tomography detected a 6 cm × 4 cm × 2.5 cm mass between the hepatoduodenal ligament and the inferior vena cava. The mass was successfully excised and the patient had a complete resolution of symptoms. A diagnosis of idiopathic unicentric CD was made based upon histological examination, which demonstrated the pathological features of CD of mixed type, including hyperplasia of follicular lymphoids with abundant plasma cells, degenerative germinal centers, interfollicular vascularity and hyaline degeneration. The diagnosis was corroborated by immunohistochemical analysis which detected multiple biomarkers.CONCLUSION This is the first study that describes the clinicopathological features of CD presenting with jaundice, which may deepen and extend our understanding of this disease.
Key words: Castleman disease; Jaundice; Case report; Clinicopathology;Immunohistochemistry
INTRODUCTION
Castleman disease (CD) is a rare lymphoproliferative disorder that was first described by Benjamin Castleman in 1956[1,2]. Its worldwide annual incidence is only around 15.9 to 19.1 cases per million subjects[3-5], but more cases have been reported recently[3]. This disease can be clinically delineated as unicentric CD (UCD) and multicentric CD(MCD) subtypes with different outcomes[6,7]. MCD occurs systemically, involves multiple groups of lymph nodes and is associated with systemic inflammatory symptoms, and the differential diagnosis is ascertained from lymphoma[8]. By contrast, UCD affects a single lymph node or a group of adjacent lymph nodes in a specific anatomical site, commonly involving the mediastinum and thoracic lymph nodes; and the mass is usually observed incidentally upon physical or medical imaging examination. In some cases, UCD is found symptomatically because of the compression of local structures by the enlarged mass. In most patients, UCD can be curatively treated by surgical excision, while systemic therapy is required for the effective management of MCD[7]. Three pathological types, namely, hyaline vascular,plasmacytic and mixed, have been reported in both UCD and MCD[7]. Here we report a rare case of UCD presenting with intermittent jaundice caused by extrinsic compression of the mass located between the hepatoduodenal ligament and the inferior vena cava. In order to extend our understanding of this disease, we review the relevant literature and summarize a total of 11 cases involving CD accompanied by jaundice.
CASE PRESENTATION
Chief complaints
A 62-year-old woman of Han ethnicity was admitted at the Fourth Affiliated Hospital of Harbin Medical University with a one-month medical history of intermittent upper abdominal pain, and skin and sclera jaundice.
History of present illness
She reported a one-month medical history of intermittent upper abdominal pain, and skin and sclera jaundice.
History of past illness
The patient did not have a specific history of past illness.
Personal and family history
Nor did her family.
Physical examination upon admission
Slight right upper quadrant abdominal tenderness was the only observed clinical sign at the time of admission.
Laboratory examinations
Serum levels of total bilirubin (53 μmol/L) and direct bilirubin (35 μmol/L) were both above normal ranges (total bilirubin: 3.1-22.5 μmol/L; direct bilirubin: 1.3-7.2 μmol/L), but declined to 23 and 12 μmol/L, respectively, one day prior to operation.All the other laboratory tests including serum levels of alpha-fetoprotein (AFP),carcinoembryonic antigen (CEA) carbohydrate antigen (CA) 19-9 and interleukin-6 were within normal ranges.
Histological examination showed follicular lymphoid hyperplasia and a shortage of lymphatic sinuses. Lymphoid follicles were mainly composed of proliferating mantle cells, and concentrically layered around highly vascularized and degenerative germinal centers (Figure 1A). Vascularity was observed in the inter-follicular regions with vessels penetrating the germinal centers (Figure 1A). Partial tissues were of hyaline degeneration (Figure 1B). Plasma cells were abundant in tissues and Russell’s bodies were sparsely distributed (Figure 1C and D).
Further immunohistochemical examination using a panel of antibodies supported the diagnosis (Figure 2). CD20+cells were mainly observed in the follicular regions and occasionally in the inter-follicular regions, corresponding to the distribution of plasma cells, since CD20 is expressed on the surface of all B-cells[9]. Paired box protein(PAX)-5+cells were observed in the follicular regions and to a lesser extent in the inter-follicular regions, since PAX-5 is expressed at early but not late stages of B-cell differentiation[10]. These results imply that B cells were at a proliferative phase. B-cell lymphoma 2 (Bcl-2) was negative in, but positive outside of, the germinal centers,indicating degeneration of germinal centers since Bcl-2 regulates cell apoptosis[11]. It has been reported that antibodies against Bcl-2 react with B-cells in the mantle zone[12].CD38+cells were mainly observed in inter-follicular regions and sparsely distributed in the germinal centers, in accordance with the abundance of plasma cells since CD38 is downregulated in B cells and mature plasma cells[13]. CD21, a marker of follicular dendritic cells[14], was positive, indicating the presence of intact follicular dendritic networks. CD3-positive T cells[15]were mainly observed in the inter-follicular regions,but only sparsely in the follicular regions. Ki-67, a cellular marker for proliferation[16],was highly expressed in 80%-90% of cells within the germinal centers but was expressed in only 5% of cells outside of the germinal centers. Immunoglobulin free light chains Kappa and Lambda-positive cells were observed in the inter-follicular regions with a ratio of 1.2:1. The expression of these two molecules indicates the relative abundance of plasma cells and could be a differential diagnosis from B-cell lymphoma, in which one type of light chain is significantly more common than the other[17].
Imaging examinations
Contrast-enhanced computed tomography (CT) of the abdomen showed a mass between the hepatoduodenal ligament and the inferior vena cava, reaching the hepatic caudate lobe and the right renal vein plane. The front side was nearby the duodenal descending segment and the pancreatic head. The mass did not have a clear boundary with the caudate lobe and the duodenum, and the portal vein was oppressed (Figure 3A-C). There was no obvious difference in the density of the mass at arterial, portal venous and equilibrium phases of CT scanning (Figure 3A-C). By using three-dimensional (3D) visualization software (Intelligent Precise Service,Shenzhen Xudong Digital Medical Imaging Technology Co. Ltd., China), a 3D visualization model was generated to display the adjacent relationships between the mass and blood vessels, the pancreas, the gallbladder and the dilated bile duct (Figure 3D and E). Apart from this mass, there was an absence of additional lesions in the rest of the body as detected by physical and imaging examinations. A preoperative diagnosis of abdominal lesion was made and the patient underwent exploratory laparotomy after an appropriate preparation. A mass was found located between the hepatoduodenal ligament and the inferior vena cava and successfully excised. The mass had an intact capsule (Figure 3F) and a clear boundary with adjacent organs including the common bile duct. The mass was 6 cm × 4 cm × 2.5 cm in size and had a greyish-white granular cutting surface with local bleeding regions (Figure 3G).
FINAL DIAGNOSIS
Based on the above clinicopathological feature and immunohistochemical analyses, a diagnosis of UCD of mixed type was made.
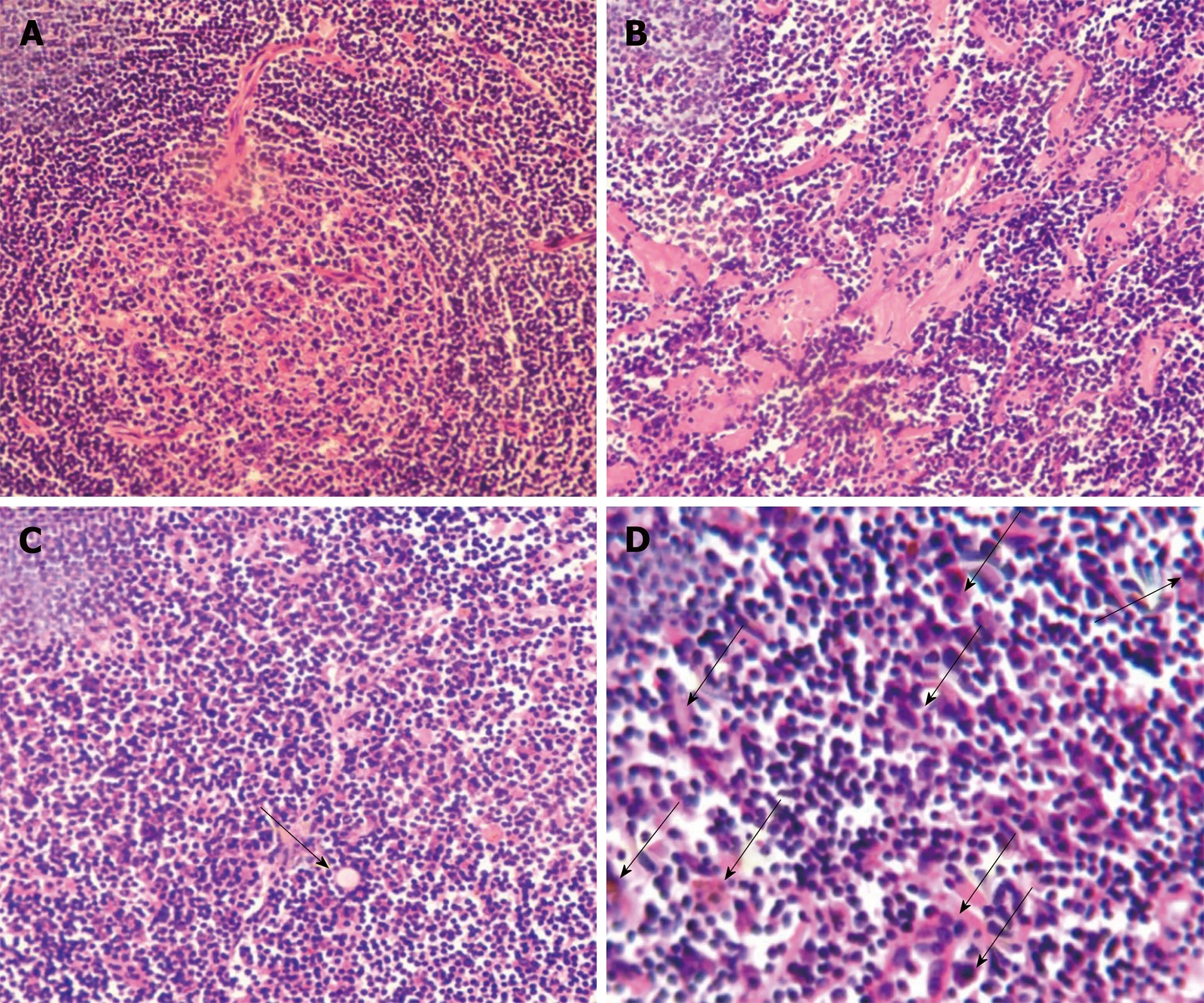
Figure 1 Major pathohistological features of mixed type Castleman disease. Tissue sections (5 μm) were stained with hematoxylin and eosin. Histological characteristics in representative images include hyperplasia of follicular lymphoids concentrically layered around vascularized and degenerative germinal centers and shortage of lymphatic sinuses (A), hyaline degeneration (B), existence of Russell’s body (arrow) (C), and abundant proliferating plasma cells (arrows) (C and D). Magnification, × 200 (A-C) and × 400 (D).
TREATMENT
As described above, the mass was surgically removed. No non-surgical treatment was given to the patient.
OUTCOME AND FOLLOW-UP
After surgical treatment, the clinical manifestations disappeared and serum levels of bilirubin returned to normal. The patient remained well without jaundice after a 12-mo follow-up.
DISCUSSION
Until now, 12 cases of CD presenting with jaundice including the present case have been described in 10 reports in the literature (Table 1)[18-27]. Of the 12 cases, MCD and UCD were equally divided; 7 (58.3%) were of hyaline vascular type, 3 (25%) of plasma cell type, and 2 (16.7%) of mixed type. All the cases of UCD were successfully excised and had a complete symptom resolution within the term of follow-up, while the cases of MCD were either partially controlled or relapsed.
CD comprises a heterogeneous group of disorders and presents with diverse symptoms and signs due to the location, size, extent and diffusion[4]. This leads to difficulties in arriving at a differential diagnosis, and surgical excision is usually not only a diagnostic but also a curative method for the management of the disease,particularly for UCD. Therefore, in addition to clinical manifestations, evaluation of CD should also include pathological analysis with immunohistochemistry, laboratory investigation and systemic imaging.
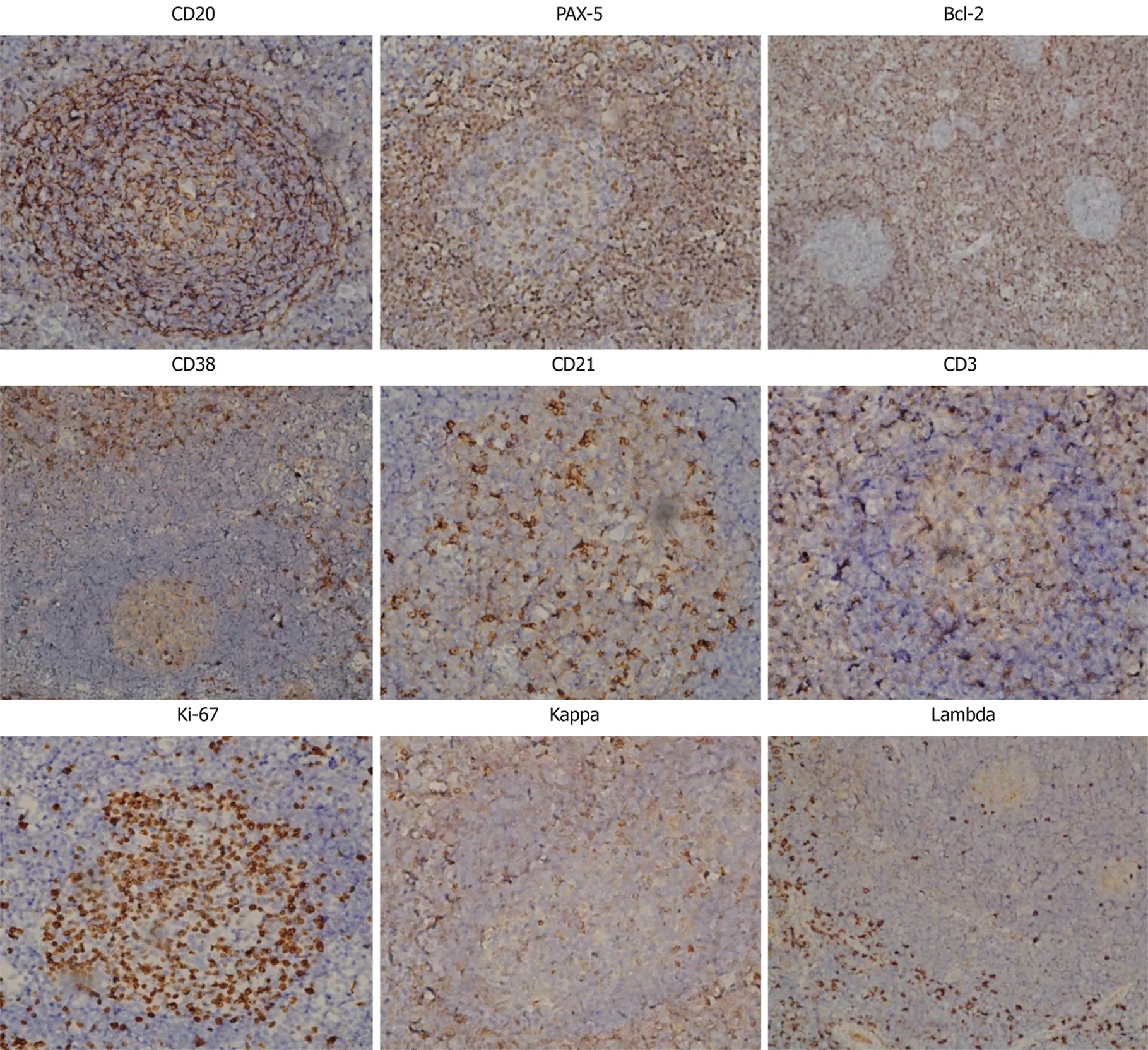
Figure 2 Immunohistochemistry. A panel of antibodies as indicated were used to immunostain the sections, which were subsequently counterstained with hematoxylin. Magnification, × 200. PAX-5: Paired box protein-5; Bcl-2: B-cell lymphoma 2.
MCD is classically distinguished from UCD by the presence of systemic symptoms,such as fever, asthenia, pleural effusion, ascites, the presence of lymph nodes in more than one region, hepatosplenomegaly, signs of inflammation and a poorer prognosis[7]. However, in approximately 60% of cases of UCD, the presentation is asymptomatic with accidental detection of a mass[28]. Some patients are diagnosed with UCD because enlarged lymph nodes impede nearby organs. The main manifestation of UCD is related to local complications of a mass with extrinsic compressions, such as renal obstruction, dyspnea, cough, abdominal pain and other signs[6]. The plasma cell variants are the most common type of MCD (75%) but represent only 10%-20% of UCD[6,28,29]. MCD may affect other organs containing lymphoid tissues and often occurs in people infected with the human immunodeficiency virus. Interleukin-6 has been shown to play a major role in the pathogenesis of both UCD and MCD since it stimulates B-cell proliferation and survival through multiple signaling pathways[9].
The majority of UCD cases are the hyaline-vascular variants, characterized by hyalinized blood vessels between follicles with few plasma cells, whereas a small number of cases are plasma cell variants, which show characteristic sheets of matureappearing plasma cells in the interfollicular region[8]. CD mimics a number of neoplastic entities and therefore immunohistochemistry can help for differential diagnosis[29]. UCD involving the pancreas and bile duct is rare, with only 11 cases reported (Table 1)[18-27]. In this case, UCD should also be differentiated from lymph node metastases of malignancies by using positron emission tomography/CT (PETCT), magnetic resonance imaging (MRI) and/or endoscopic retrograde cholangiopancreatography (ERCP), which would guide the therapeutic management.An endoscopic ultrasound-guided fine needle aspiration biopsy (EUS-FNA) would also help to make a pathological diagnosis preoperatively. Unfortunately, none of the above examinations apart from dynamic CT were employed in this case due to the inaccessibility of facilities and high cost. Radiation therapy may be an option for the disease with incomplete excision, or if the patient is in poor health and unable to undergo surgery.
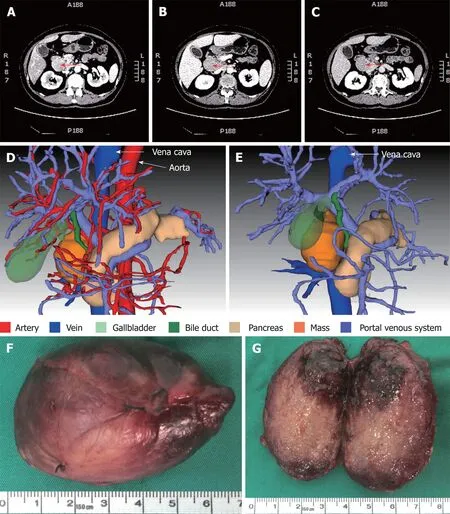
Figure 3 Computed tomography scan, three-dimensional visualization model and the mass. A-C: Representative images are from enhanced multi-phase abdominal computed tomography scans at the arterial (A), portal venous (B) and equilibrium (C) phases. Arrows point to the mass. D and E: Three-dimensional images were generated to show the adjacent relationship between the mass and adjacent organs and tissues. F: The excised mass. G: The cutting surface.
In summary, here we report a case of CD presenting with intermittent jaundice with slight abdominal tenderness. Contrast-enhanced CT scanning detected a mass between the hepatoduodenal ligament and the inferior vena cava, which was successfully excised, and the patient had a complete symptom resolution. Histological examination demonstrated the pathological features of idiopathic UCD of mixed type,supported by immunohistochemical detection of multiple markers. Excluding the current case, there have been only 11 reported cases of CD presenting with jaundice.To our knowledge, this is the first study that combines clinicopathology and immunohistochemistry in diagnosing CD accompanied with jaundice, which may deepen and extend our understanding of this orphan disease.
CONCLUSION
The case of CD with jaundice presented herein, together with literature review,indicates that CD may present with diverse clinical symptoms and signs because of the location, size, extent and diffusion of the mass. The information from clinical symptoms and signs, laboratory tests, imaging examinations, pathological and immunohistochemical analyses should be comprehensively considered. Otherexaminations not used in the present case, such as PET-CT, MRI, ERCP and EUSFNA, may be helpful for an accurate preoperative diagnosis and therapeutic management.
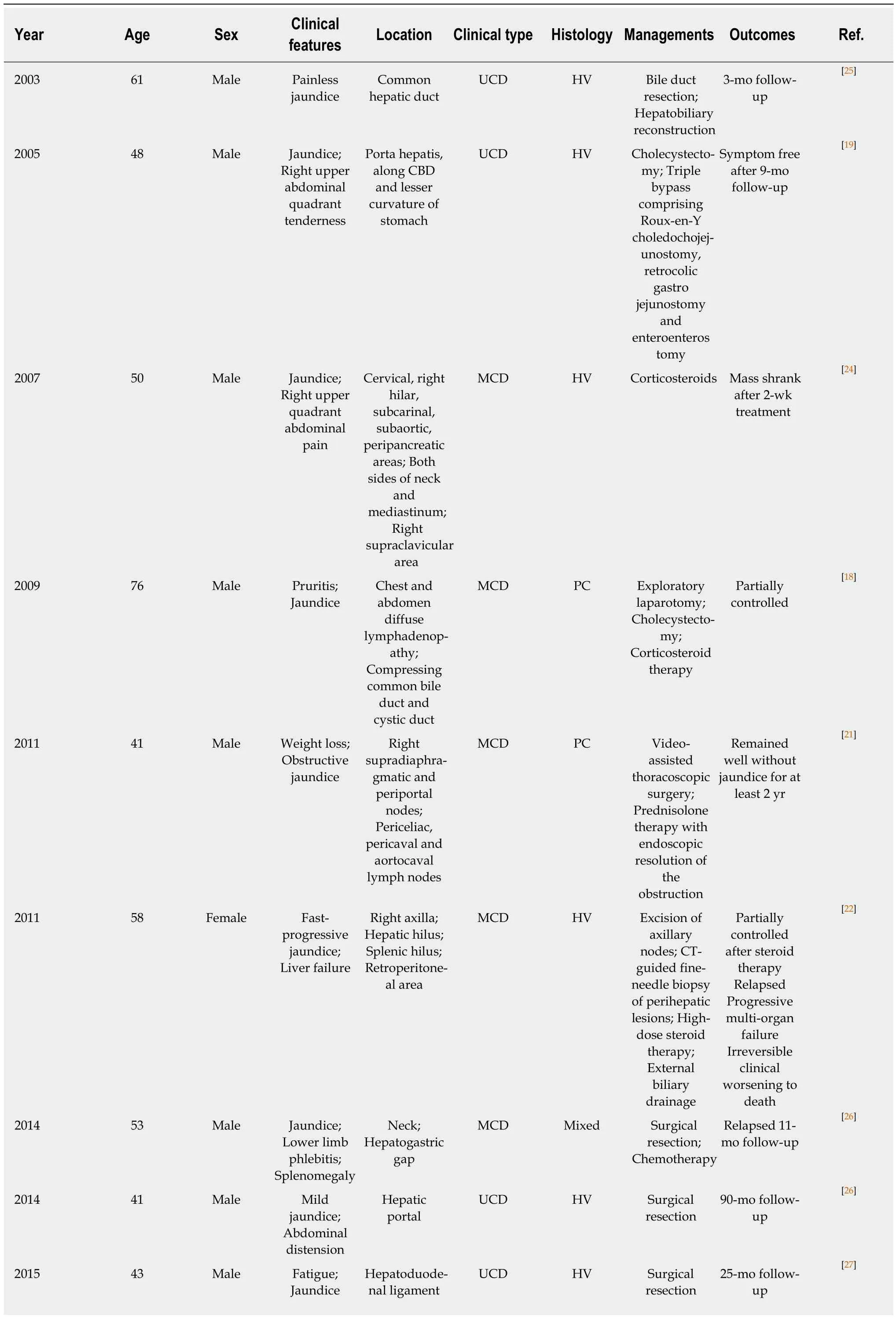
Table 1 Summary of reported cases of Castleman disease presenting with jaundice
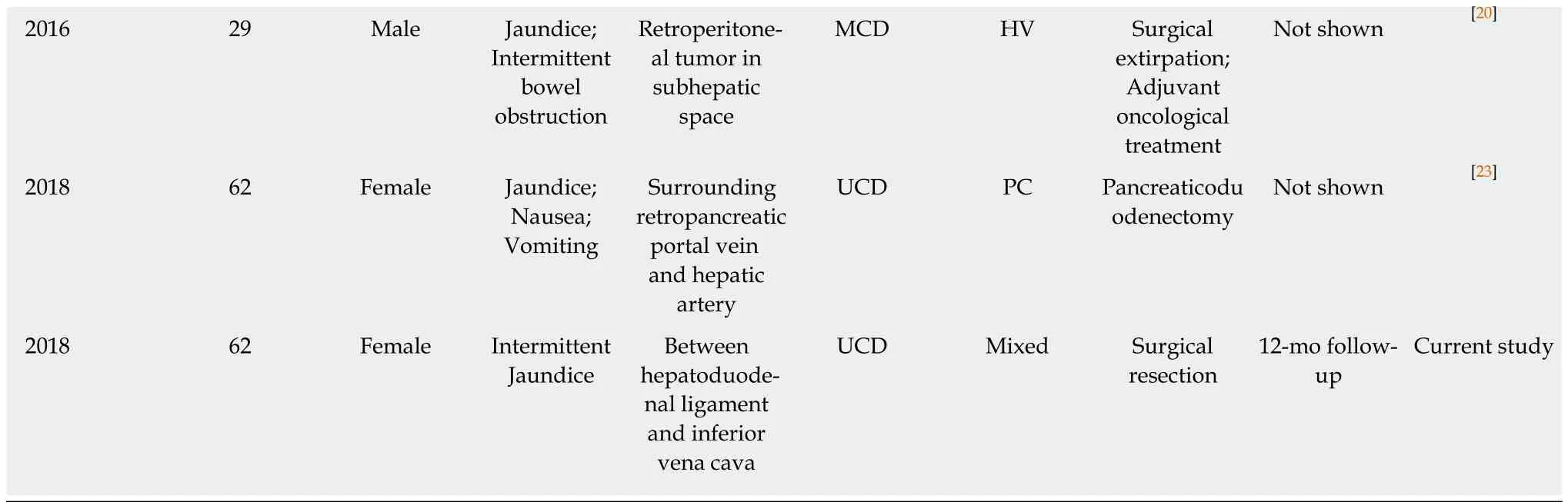
UCD: Unicentric Castleman disease; MCD: Multicentric Castleman disease; HV: Hyaline vascular; PC: Plasmacytic; CT: Computed tomography.
 World Journal of Clinical Cases2019年3期
World Journal of Clinical Cases2019年3期
- World Journal of Clinical Cases的其它文章
- Biventricular pacing for treating heart failure in children: A case report and review of the literature
- Gerstmann-Sträussler-Scheinker disease: A case report
- Considerations for routine coagulation monitoring with rivaroxaban:A case report and review of the literature
- Papillary cystadenoma of the parotid gland: A case report
- Ghost cell odontogenic carcinoma of the jaws: Report of two cases and a literature review
- Intravenous leiomyomatosis with different surgical approaches:Three case reports