
改良比色法检测食品中的亚硝酸盐
2019-02-20 02:58:32苗攀登刘钟栋
食品工业科技 2019年23期
苗攀登,刘钟栋
(河南工业大学粮油食品学院,河南郑州 450001)
亚硝酸盐是一种很常见的食品添加剂和天然衍生物[1],在香肠、果酱、腌制品、肉脯等食品中都有不同剂量的添加,主要是起着色和防腐的作用。亚硝酸盐是剧毒物质,其成人中毒剂量为0.2~0.5 g,致死剂量为3 g。在胃酸等环境下亚硝酸盐与食物中的仲胺、叔胺和酰胺等反应生成强致癌物N-亚硝胺,它有极强的致癌作用。亚硝酸盐还能使血液中正常携氧的低铁血红蛋白氧化成高铁血红蛋白,因而失去携氧能力而引起组织缺氧。此外,亚硝胺还能够透过胎盘进入胎儿体内,对胎儿有致畸作用。6个月以内的婴儿对亚硝酸盐特别敏感,临床上患“高铁血红蛋白症”的婴儿即是食用亚硝酸盐浓度高的食品引起的,症状为缺氧,出现紫绀,甚至死亡[2-4]。
食品在生产、销售等过程中,均存在着各种各样的安全隐患,其中亚硝酸盐超标是一个非常严重的问题。亚硝酸盐的危害无需赘述,所以详细记录低值食品原料中的亚硝酸盐含量的变化,是控制亚硝酸盐含量的必要手段。现阶段的检测手段有光度法、化学发光法、电化学法、色谱法、毛细管电泳法。最常见的光度法即是比色法,其原理是:在酸性环境下,对氨基苯磺酸首先与亚硝酸根反应生成重氮化合物,然后再与N-(1-萘基)-乙二胺发生偶合反应,形成紫红色偶氮化合物,在540 nm波长处具有最大吸收且与亚硝酸盐含量呈正相关,检测限为0.06 μg/mL。此方法已经非常成熟、重复性好,准确度高,但步骤繁杂,易受其它杂质干扰,试剂毒性较大,也鉴于其检测限的限制,近些年逐步被离子色谱法取代[5-9]。化学发光法的原理是根据亚硝酸盐浓度与分子发光强度呈线性关系,通过检测分子发光强度来得到亚硝酸盐的浓度。此方法具有操作方便灵敏度高、试剂消耗少、仪器设备简单等优点,但也有实验仪器昂贵、操作人员专业要求高、工作环境要求高等缺陷[10-11]。电化学方法中常见的极谱法,是利用电解过程中所得到的极化电极的电流电位曲线来确定溶液中物质浓度的电化学分析方法,具有设备简单、分析速度快、准确度高、灵敏度高的优点,但是有设备昂贵、不易保养以及重现性差等缺陷[12-13]。离子色谱法是利用阴阳离子对载体(树脂)的吸附能力不同达到分离的目的[14-16]。总的来说,色谱法具有线性范围宽、抗干扰能力强、操作简便特点,但有设备仪器昂贵、操作人员要求高、耗时等缺点;此外,亚硝酸盐的检测方法还有毛细管电泳法、催化光度法、荧光光度法、流动注射分光光度法、电化学伏安法、气相色谱法以及高效液相色谱法等。光度法使用方便但是不够灵敏,容易受干扰,其他方法虽有达到较低检测限,但是设备昂贵,不易保养,操作繁杂,不宜大批检测样品,又或者耗时长,时效性不高。因此改进光度法,使其克服固有的缺点是一个建立新方法的思路。
在本研究中,将对氨基苯硫酚(ATP)和萘基乙二胺(NED)分别修饰在纳米金颗粒上之后,在酸性条件下,再加入亚硝酸盐,体系将会发生重氮化耦合反应,使金纳米颗粒发生聚集,从而导致体系光学性质(颜色)发生变化。通过探究颜色变化与亚硝酸盐浓度的关系来建立亚硝酸盐检测方法,计算检测限。
1 材料与方法
1.1 材料与仪器
面包 包头市味多美食品有限公司;火腿 河南双汇投资发展股份有限公司;海带丝 山东烟台万历海藻食品有限公司;袋装牛奶 内蒙古伊利实业集团股份有限公司;瓶装水 康师傅控股有限公司;实验所用食品材料 为市场随机购买所得;水合氯金酸(HAuCl4) Sigma Aldrich公司;柠檬酸钠、对氨基苯磺酸、对氨基苯硫酚ATP、盐酸萘乙二胺NED、巯基乙酸 北京化学试剂公司;亚硝酸钠标准溶液(1000 μg/mL) 成都博瑞特化学技术有限公司,可直接使用无需校准;浓盐酸、氢氧化钠 天津市大茂化学试剂厂;无水乙醇、亚铁氰化钾、乙酸锌 麦克林试剂有限公司;去离子水 天津卓精虹科仪器设备技术有限公司;其他化学试剂 均为分析纯。
U-2900型紫外分光光度计 Hitachi;ZS90型粒度电位仪 Malvern;琼脂糖电泳设备 天津市华茂电泳设备有限公司;磁力搅拌器及其配套电热套 巩义市予华仪器设备有限公司;小型振荡器 上海亚荣生化仪器厂;离心机 上海卢湘仪;JR05-300型食品粉碎机 浙江苏泊尔股份有限公司;KQ-100DE型超声波仪(输出功率:400 W)昆山市超声仪器有限公司以及其他常规实验室器皿。
1.2 实验方法
1.2.1 样品前处理方法 取面包、火腿、海带丝各0.25 g分别放入食品粉碎机再加入10 mL水,粉碎25 min,并转移到25 mL容量瓶中,三次用少量水(2 mL左右)清洗粉碎机杯壁倒入容量瓶中,再定容到25 mL;瓶装水和袋装牛奶直接转移到容量瓶并定容到25 mL[17-19]。所有样品定容后,在超声清洗机中(400 W)浸泡30 min,然后2000 r/min离心15 min,取上清液5 mL(其中瓶装水样品无分层),加入200 μL亚铁氰化钾溶液(106 g/L),再加入0.5 mL乙酸锌溶液(220 g/L),手动搅拌5~10 min,过滤得到样品液[20];用体系检测样品液亚硝酸盐浓度c(μg/mL),即样品的亚硝酸盐含量:C=c×25/0.25(μg/g),C=100c。
1.2.2 传统比色法检测亚硝酸盐
1.2.2.1 传统比色法的标准曲线的绘制 参考国标GB 5009.33-2016,1000 μg/mL的亚硝酸钠标准液用去离子水分别稀释到0.05、0.1、0.5、1.0、3.0、5.0、10.0、15.0、20.0 μg/mL,再分别取4.7 mL稀释液和去离子水(空白),分别放在试管中,加入0.2 mL 4 g/L对氨基苯磺酸溶液,混匀,静置3~5 min后各加入1 mL 2 g/L盐酸萘乙二胺溶液,混匀静置10 min。用1 cm比色杯,以空白管调节零点,于波长538 nm处测吸光度,绘制标准曲线[21]。

1.2.2.3 传统比色法的加标回收试验 以正常销售的鲜牛奶和瓶装水为空白,分别取鲜牛奶和瓶装水2.5 g。向两种样品中分别加入1.00、5.00、10.00、20.00 μg/mL的亚硝酸盐标准品,用去离子水定容25 mL,在输出功率400 W的超声仪中浸泡30 min;离心取上清液5 mL,加入200 μL亚铁氰化钾溶液(106 g/L),再加入0.5 mL乙酸锌溶液(220 g/L),搅拌5~10 min,过滤得到样品液,然后用传统比色法检测样品液的亚硝酸盐浓度c,即亚硝酸盐的添加量m=c×25,计算出回收率并重复10次计算相对标准偏差。
1.2.3 改良比色法检测亚硝酸盐
1.2.3.1 实验原理 利用金纳米颗粒优秀的光学性质,将对氨基苯硫酚通过S-Au键修饰到水相金纳米颗粒记为ATP-GNPs;将萘基乙二胺(NED)通过1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDC)催化,连接到双亲性聚合物包裹的油相金纳米颗粒(AP-GNPs)上得到萘基乙二胺修饰的金纳米颗粒记为NA-GNPs,在酸性条件下,加入NO2-后,发生重氮化耦合反应,两种修饰过的金纳米颗粒被连在一起,发生聚合,金纳米颗粒的颜色发生明显变化,这种变化与NO2-的浓度呈良好线性关系。总之,改进比色法用金纳米颗粒的灵敏的光学变化代替了耦合反应生成的染料分子的颜色变化,使体系的检测线更低,灵敏度更高,如图1。

图1 改良比色法基本原理示意图Fig.1 Schematic diagram of the the improved colorimetric basic principle
1.2.3.2 ATP-GNP的制备及表征 取17 mg水合氯金酸溶于50 mL的去离子水中,沸腾回流10 min,溶液呈淡黄色;78 mg柠檬酸钠溶于5 mL去离子水中,预热到60~70 ℃,快速加入到回流后的水合氯金酸的溶液中,继续加热回流20 min,溶液颜色由黄色变无色随后颜色逐渐加深,最后呈透亮的深红色,冷却至室温,调节pH到7,用0.45 μm的滤膜过滤两次,室温备用,这时的金纳米颗粒GNPs的粒径大约为10 nm[22]。由郎伯比尔定律(式1)计算得到的GNPs的吸光度为A=1.924,可计算得知GNPs的浓度c=1.40×10-8mol/L。
郎伯比尔定律A=ε×C×l
式(1)
式中:A是吸光度值,ε是吸收常数,l是光径长度(cm,所使用的仪器光径长度规格为1 cm),ε10 nm=1.37×108cm-1mol-1L。
将制备好的金纳米颗粒稀释10倍,将pH调整到弱酸性(pH=3~4),用10 mmol/L的ATP(溶解于无水乙醇)与稀释后的纳米金颗粒按体积1∶8的比例混合均匀,在摇床上匀速振荡6 h,反应后,用5000 r/min离心30 min,去上清,再用去离子水溶解,反复3~5次,即得到ATP-GNPs。ATP-GNPs的粒径约为10 nm,由郎伯比尔定律[23-24],计算出ATP-GNPs浓度约为10-9mol/L。
制备ATP-GNPs之后,在室温下利用粒径电位仪、全波谱紫外分光光度计表征GNPs和ATP-GNPs的粒径分布、电位变化以及吸收图谱的变化,其中样品皿的规格为:1×1 cm的石英皿。
1.2.3.3 NA-GNPs的制备及表征 有机相金纳米颗粒是在有机相中用硼氢化钠快速还原氯金酸得到的,再利用两亲性的聚合物的包裹使其“溶解”到水相中,再通过反应将萘基乙二胺连接到两亲性聚合物的羧基上,最终得到萘基乙二胺修饰的金纳米颗粒(NA-GNPs),详细反应步骤见a~d。
a:有机相合成金纳米颗粒GNPs:取2.17 g四辛基溴化铵溶于80 mL甲苯中,在输出功率400 W的超声仪中浸泡2~3 min使四辛基溴化铵完全溶解,溶液澄清透明无杂质[25]。取300 mg氯金酸溶于25 mL纯水中,溶液为澄清的黄色。上述两种溶液混合,轻摇直到水相中的黄色完全消失,有机相变成红色。用分液漏斗将水油分开,有机相转移到烧瓶中。334 mg硼氢化钠溶于25 mL纯水。硼氢化钠溶液在1 min之内加入之前含有金的甲苯溶液中,边揽拌边滴加。然后金溶液在室温下搅拌l h。搅拌l h之后,再利用分液漏斗溶液将水油分开,有机相先用10 mmol/L盐酸洗3遍,再用10 mmol/L氢氧化钠洗3遍。将溶液转到烧瓶中搅拌12 h后加入10 mL十二硫醇,65 ℃水浴搅拌3 h。所得溶液2000 r/min离心5 min,收集上清,去除杂质。再按1∶1加入甲醇2000 r/min离心5 min。去除含有四辛基溴化铵和十二硫醇的上清液,收集GNPs沉淀溶于氯仿中。合成的GNPs粒径约5 nm。
b:制备两亲性聚合物:2.7 g十二胺和3.084 g聚(异丁烯-alt-马来酸酐)溶于100 mL四氢呋喃,55~60 ℃水浴搅拌l h。此过程四氢呋喃会挥发,溶液大概浓缩到30~40 mL。停止加热,剩余溶液在室温下搅拌12 h。利用真空旋转蒸发仪使四氢呋喃完全挥发,加入40 mL氯仿剧烈搅拌,使瓶底附着的物质完全溶解,最终所得两亲性聚合物AP。
c:AP包被GNPs:取1 mL GNPs(溶于氯仿)于圆底烧瓶,再加入0.2 mL AP,摇晃均匀,加入5 mL的氯仿降低体系浓度。缓慢抽真空使氯仿挥发直到混合物完全干燥。加入适量SB12(0.05 mol/L硼酸缓冲液,pH=12)剧烈搅拌,使粘附在瓶壁上的物质完全溶解。用60000 r/min离心30 min进行纯化,去上清用去离子水复溶,重复3~5次,就得到两亲性聚合物包被的金纳米颗粒,记为AP-GNPs,如图2。

图2 AP包被GNPs的示意图Fig.2 Scheme of AP coating on GNPs
d:萘基乙二胺修饰AP-GNPs得到NA-GNPs:10 mL 20 mmol/L盐酸萘乙二胺溶液(溶于硼酸缓冲液,pH调到9)与1 mL AP-GNPs混匀,再加入1 mL 0.5 mol/L的EDC,混匀静置3 h。用60000 r/min离心30 min进行纯化,去上清用去离子水复溶,重复3~5次,就得到NA-GNPs,其粒径约为10 nm。由郎伯比尔定律,计算出NA-GNPs浓度约为210-7mol/L。将NA-GNPs稀释20倍后,与ATP-GNPs等体积混合,将体系pH调整到5,作为反应液存放于-4 ℃备用。将亚硝酸钠标准溶液分别稀释到0.005、0.01、0.05、0.1、0.5、1、2、3、4、5 μg/mL。分别取浓度梯度的标准液和去离子水(空白)0.5 mL,再加入0.5 mL反应液,混匀静置15~20 min,测体系的吸光度,绘制标准曲线。
e:表征:在室温下利用粒径电位仪、全波谱紫外分光光度计表征油相GNPs、AP-GNPs和NA-GNP的粒径分布、电位变化以及吸收图谱的变化,其中样品皿的规格为:1×1 cm的石英皿。
1.2.4 存放时间和其他离子对检测体系的影响 将反应液存放1、2、3、4、5周,分别测紫外吸光度,观察金纳米颗粒520 nm处的特征吸收峰,证明反应液的稳定性。
1.2.5 改良比色法的加标回收试验 以正常市售的鲜牛奶和瓶装水为空白,分别取鲜牛奶和瓶装水2.5 g。向两种样品中分别加入5、10、20 μg的亚硝酸盐标准品,定容25 mL,搅拌超声浸泡30 min;以2000 r/min离心15 min取上清液5 mL,加入200 μL亚铁氰化钾溶液(106 g/L),再加入0.5 mL乙酸锌溶液(220 g/L),搅拌5~10 min,过滤得到样品液,然后用改良比色法检测样品液的亚硝酸盐浓度c,即亚硝酸盐的添加量m=c×25,计算出回收率并重复10次计算相对标准偏差[26-27]。
1.2.6 两种比色法的实际应用 将面包、火腿、海带丝打开包装,放置在常温(25 ℃)下0、5、10、15、20、25、30、35、40 h,分别取0.25 g各个时段的面包、火腿、海带丝,按照1.2.1制备样品液,C样品=100c样品液(μg/g)。用传统比色法和改良比色法检测样品液,对比两种比色法的差异。
2 结果与分析
2.1 传统比色法检测亚硝酸盐
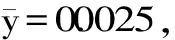
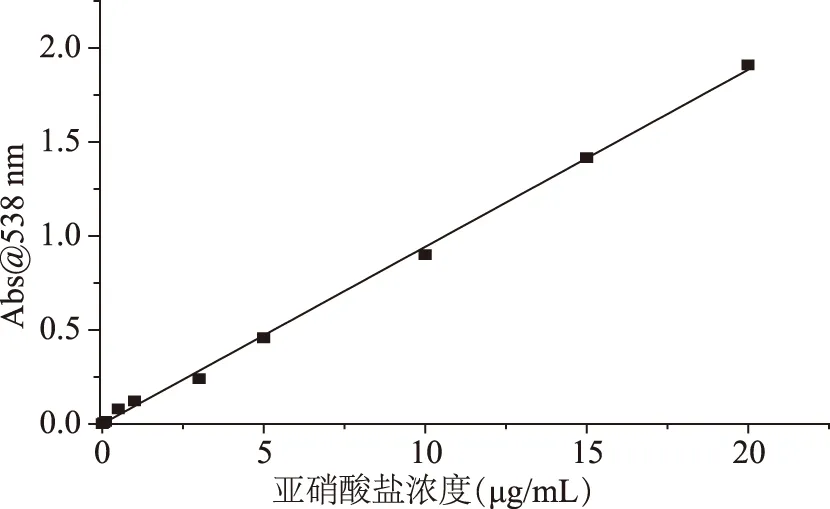
图3 传统比色法的标准曲线和各样品的颜色变化Fig.3 Standard curve of traditional colorimetric method and the color changes of the samples


图4 不同离子对体系的影响Fig.4 Effect of different ions on the reaction solution
2.1.3 传统比色法的加标回收试验 按照1.2.2.3的方法,传统比色法的加标回收试验结果如表1。由表1可见,在加入1.00 μg/mL时,传统比色法无法检测出准确的亚硝酸盐含量,显然加标样品的亚硝酸盐含量已经低于其检测限,因此传统比色法在应用方面有检测限的局限。在5.00、10.00、20.00 μg/mL的加标样品的回收率为97.88%~100.24%,相对标准偏差(RSD)为3.53%~7.22%。
表1 传统比色法的回收率以及相对标准偏差Table 1 Recovery rate and relative deviation(RSD)of the traditional colorimetric method(n=10)
2.2 ATP-GNPs的表征
通过1.2.3.2的方法制备出水相金纳米颗粒(GNPs),对氨基苯硫酚(ATP)修饰到金纳米颗粒之后,由于金纳米颗粒表面配体的变化,紫外吸收峰有少许偏移,纯化过程有少量损失,所以吸收峰强度下降,如图5;表面电位也有变化,如图6;ATP-GNPs的粒径约是10 nm,如图7。

图5 ATP修饰GNPs前后紫外吸收图谱的变化Fig.5 Changes in UV absorption spectra before and after ATP-modified GNPs

图6 ATP修饰GNPs前后表面电位的变化Fig.6 Changes in surface potential before and after ATP-modified GNPs

图7 ATP-GNPs的粒径表征Fig.7 Particle size characterization of ATP-GNPs
2.3 油相GNPs、AP-GNPs、NA-GNPs的表征
用1.2.3.3方法步骤制作出油相GNPs的粒径约为5 nm,用AP包被后的AP-GNPs的粒径约为10 nm,而且紫外吸收吸收峰发生偏移;由于疏水性,油相GNPs被AP包被,亲水的羧基暴露在外侧,在EDC的催化下与萘基乙二胺(NED)的氨基连接在一起。纯化后把多余的NED去除,就得到了NA-GNPs,由于NED的分子较小,因此NA-GNPs的紫外吸收峰相对于AP-GNPs几乎没有变化,粒径的变化也不大,只有纯化过程中有损失,峰强度降低。油相GNPs、AP-GNPs、NA-GNPs的紫外吸收图谱、粒径,如图8、图9。
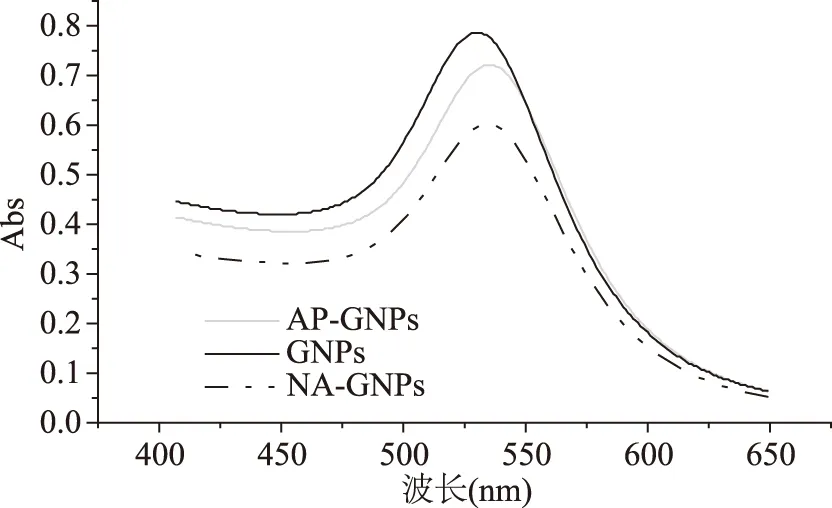
图8 油相GNPs、AP-GNPs、NA-GNPs的紫外吸收图谱Fig.8 Ultraviolet absorption spectra of oil phase GNPs,AP-GNPs and NA-GNPs
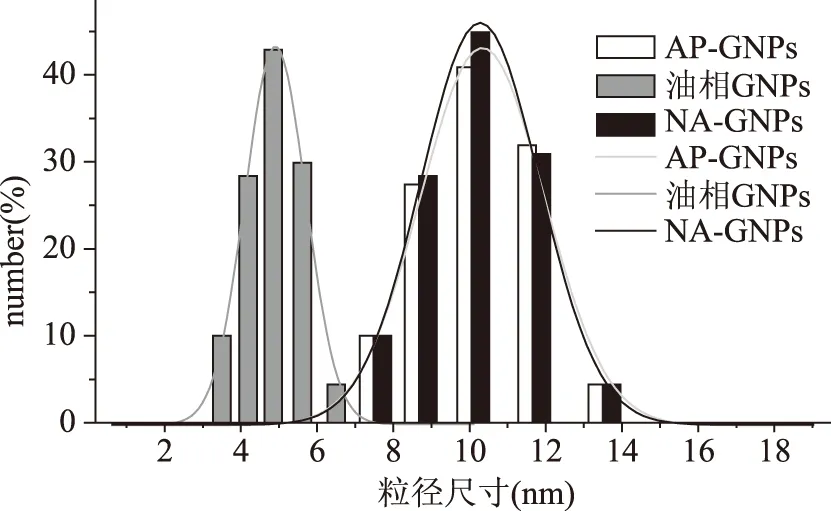
图9 油相GNPs、AP-GNPs、NA-GNPs的粒径表征Fig.9 Size characterization of oil phase GNPs,AP-GNPs and NA-GNPs
2.4 改良比色法的结果分析


图10 加入浓度梯度溶液后体系的紫外吸收图谱Fig.10 UV absorption spectrum of the system by adding a concentration gradient solution

图11 浓度梯度的的标准曲线Fig.11 Standard curve of concentration gradient
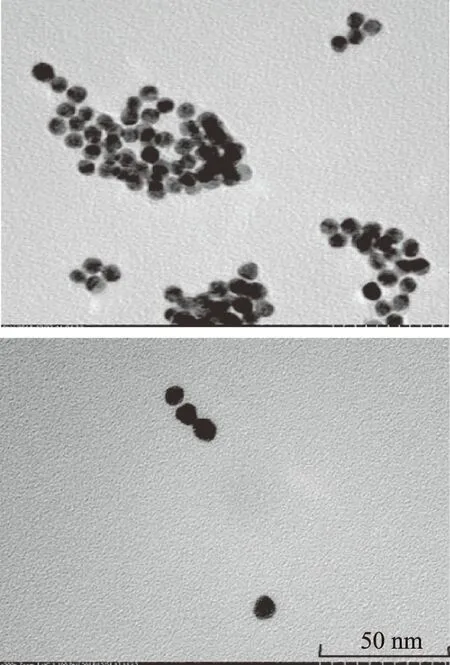
图12 同等条件下,加入溶液前(右)和后(左),金纳米颗粒的聚集情况Fig.12 Aggregation of gold nanoparticles before(right)and after(left)addition of solution under equivalent conditions
利用空白样品液10次测量吸光度得到10个A580 nm/A520 nm的值,其标准偏差为0.11%。由公式dl=3σ/k,其中dl是检测限,σ是指空白样品的标准偏差,k是指标准曲线的斜率。通过计算体系的检测限dl=5.12 ng/mL。
2.5 存放时间和其他离子对检测体系的影响

图13 不同存放时间,反应液的紫外吸收图谱Fig.13 UV absorption spectrum of the reaction solution at different storage time

图14 不同离子对反应液的影响Fig.14 Effect of different ions on the reaction solution
2.6 改良比色法的加标回收试验
按照1.2.5的方法,改良比色法的加标回收实验结果如表2。
由表2可见,样品回收率为97.00%~100.28%,相对标准偏差(RSD)为0.77%~3.88%,表明本实验方法具有可靠地准确度和精密度。
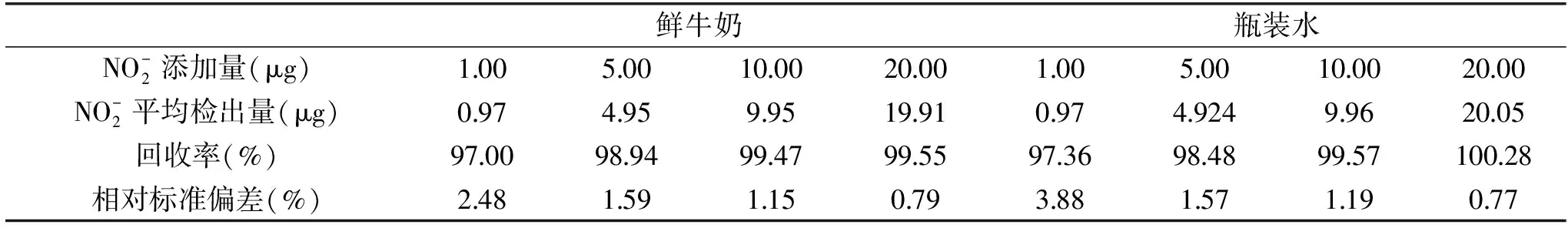
表2 改良比色法的回收率以及相对标准偏差Table 2 Spike recovery rate and relative deviation(RSD)of the improved colorimetric method(n=10)
2.7 改良比色法与传统比色法的比较
2.7.1 检测范围和检出限的比较 两种方法的检测范围差异很大,表明了改良比色法检测极微量的亚硝酸盐方面的优势,但是传统比色法检测范围更广。改良比色法的检测限低至5.12 ng/mL,比传统比色法灵敏度更高。在2.6中也能得到证实,加标1.00 μg的样品处理液的浓度为40 ng/mL,传统比色法无检测信号,然而改良比色法能够正常的检测其浓度,因此显示出改良比色法的优势。
2.7.2 回收率的比较 改良比色法和传统比色法各自的检测范围内的回收率相差无几,约为97%~100%,说明两种方法在各自的检测范围内都有良好的准确性和可靠性;传统比色法的RSD为3.53%~7.22%,明显比改良比色法大,说明改良比色法检测结果的离散度程度较小,即检测稳定性比传统比色法优秀。
2.7.3 响应时间的比较 传统比色法是先用对氨基苯磺酸与亚硝酸根发生重氮化反应(3~5 min),再与萘基乙二胺盐酸盐发生耦合显色(15 min),整个过程需要15~20 min,而改良比色法的对氨基苯硫酚和萘基乙二胺连接到金纳米颗粒后,使用时只加入亚硝酸钠溶液即可,这一过程约15 min,所以改良比色法操作简单,节约反应时间,提高效率。

表3 改良比色法与传统比色法的比较Table 3 Comparison between improved colorimetry and traditional colorimetry
2.7.4 稳定性的比较 改良比色法的两种金纳米颗粒混合后可以稳定存放5周以上,Cu2+、Fe2+、Fe3+对新旧两种检测体系都有少许影响,可能是因为较高浓度的干扰离子本身的颜色对检测体系的吸光度有一些影响;总之改良比色法具有良好的稳定性和抗离子干扰的能力。
2.7.5 相对标准偏差RSD的比较 传统比色法的RSD为3.53~7.22,明显比改良比色法大,说明改良比色法检测结果的离散度程度较小,即检测稳定性比传统比色法优良。
总的来说,与传统比色法相比,改良比色法具有检测限低、灵敏度高、操作简单、检测效率高、稳定性好等优势,但是检测范围窄是改良比色法的缺点。改良比色法适用于极微量的亚硝酸盐的检测,如饮用水、牛奶、婴儿食品等亚硝酸盐含量允许添加量极低的食品的检测,或者研究食品中亚硝酸盐的产生过程。
2.8 两种比色法的实际应用
查阅中国的食品标准,面包的允许残留量为2 g/mg,火腿的允许残留量为70 μg/mg,海带丝的允许残留量为20 μg/mg。如图15,面包的亚硝酸盐含量较低,传统比色法检测时,30 h之前无法响应,只有亚硝酸盐增加到传统比色法的检测限后,才能有响应。然而改良比色法检测样品时,能够很好地反应亚硝酸盐的增长过程;火腿的亚硝酸盐含量较高,如图16,20 h后,样品的亚硝酸盐含量就达到了新方法的检测极限,于是体系处于饱和状态,无法准确记录后面的样品。火腿中亚硝酸盐含量较高,而且暴露15 h后,亚硝酸盐增速加大,25 h左右就超过了允许残留量,如图17。海带丝在工厂腌制过程中,一般都存放超过20 d,产品中有一定的乳酸菌消耗了亚硝酸盐,因此样品的亚硝酸盐含量增速较慢,但是也不宜存放超过30 h。总之,改良比色法优化了检测限,但是检测范围较窄(0~1 μg/mL),因此改良比色法适用于极低含量亚硝酸盐的测定,如奶制品、面制品等;另一方面,改良比色法可以用于环境污染监测,如水库、江河水的亚硝酸盐含量的检测等。
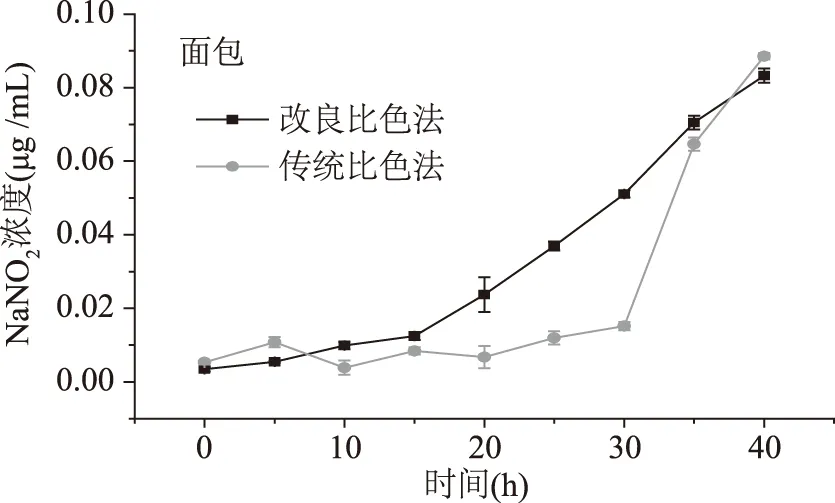
图15 面包中的亚硝酸盐的含量变化Fig.15 Changes in nitrite content in bread

图16 火腿中的亚硝酸盐的含量变化Fig.16 Changes in nitrite content in ham

图17 海带丝中的亚硝酸盐的含量变化Fig.17 Changes in nitrite content in seaweed strips
3 结论
在本研究中,利用功能化金纳米颗粒对传统比色法进行了改良。相比传统比色法,改良比色法的检测限达到了5.12 ng/mL,远远低于传统比色法(76.43 ng/mL)。改良比色法的操作步骤简便、响应时间短,大大提高了工作效率。虽然Cu2+、Fe2+、Fe3+对传统比色法和改良比色法都有影响,但是改良比色法在回收率、RSD方面有很大优势,说明此方法的稳定性更高,更能适应更复杂的工作环境。改良比色法的检测范围较窄,更加适用于微量亚硝酸盐含量的检测,是传统比色法的有效补充和改进。在应用中发现,食品打开包装后,应尽快用完,否则有亚硝酸盐超标的危险。改良比色法具有高灵敏性的特点,有利于研究亚硝酸盐产生的过程和原因,以及预防亚硝酸盐的产生。
权威·高效·核心·领先·精湛·实用
猜你喜欢
石油沥青(2023年4期)2023-09-21 07:18:12
含能材料(2022年4期)2022-04-16 06:28:04
现代畜牧科技(2021年6期)2021-07-16 05:50:28
当代水产(2019年6期)2019-07-25 07:52:16
当代水产(2018年12期)2018-05-16 02:49:52
兽医导刊(2016年12期)2016-05-17 03:51:46
安徽地质(2016年4期)2016-02-27 06:18:20
中国粮油学报(2016年1期)2016-02-06 02:17:11
应用化工(2014年1期)2014-08-16 13:34:08
中国药业(2014年24期)2014-05-26 09:00:15
