
一氧化碳在钴表面活化的轨道相互作用解析
2019-02-15 11:21杨云涛吴艳波
山西大学学报(自然科学版) 2019年1期
杨云涛,吴艳波
(山西大学 分子科学研究所,山西 太原 030006)
0 引言
理解吸附物与金属催化剂的相互作用对非均相催化反应非常重要,这不仅能够帮助理解吸附物在反应中的作用,还能用于探索金属催化剂的表面结构和功能[1]。其中CO作为吸附物与金属表面的相互作用引起了许多科学家的兴趣[2-3]。比如对费托合成反应的研究,其中CO的吸附和活化是初始反应[4-6],关于反应机理的研究已经有了很多成果。但CO中C≡O三键是如何解离的,有很多不同的观点[7]。在1926年,Fischer 和 Tropsch提出碳化物机理(Carbide mechanism),认为吸附的CO在金属表面直接解离,形成金属碳化物和氧化物,烃链的增长是通过氢化金属碳化物得到的。后续大量的理论与实验研究结果支持该机理[8-9]。但在1951年Storch提出了烯醇机理(enol mechanism)和1981年Fahey提出了甲醛机理(formaldehyde mechanism),两种机理都是在C≡O三键上吸附氢之后,生成了C-OH(醇)或H-CO(醛)等表面吸附物后发生解离,同样也有大量的理论与实验研究结果证明CO是经过氢助之后再解离[10-12]。虽然当前的研究已经取得了一定的成果。但是CO解离机理始终存在争论。这与目前的研究方式有关,目前的研究主要是选取合适的结构模型[13]、尺寸[14]或覆盖度[15]等,通过比较反应能垒的高低来确定反应进行的难易程度,很少从电子结构的角度对反应进行探讨,这使得人们对反应发生实质的理解不够深入。
在1964年[16],Blyholder从前线分子轨道的角度提出Blyholder 模型来描述CO与金属的相互作用,如图1所示,即5σ-2π*给予-反馈作用(5σ-donation/2π*-back-donation),当时对CO变化的解释主要考虑的是整个吸附物的π系统,而假设CO分子的5σ轨道未受影响。之后,Hammer[17],Nilsson[18],Bennich[19],Föhlisch[20]等人提出另一种解释:π-σ吸引-排斥机制(π-attraction σ-repulsion),他们认为杂化轨道dπ是由CO的1π和2π*与金属的dxz,yz相互杂化得到,CO的成键强度被削弱是因为在杂化轨道1π上碳被极化,形成π吸引作用,该解释认为金属对CO的反馈是非直接的dxz,yz→2π*。而杂化轨道dσ是CO的5σ与dz2杂化得到,通过电子重新分配形成的杂化σ轨道抑制了杂化轨道1π上碳的极化作用,产生σ排斥作用,π吸引和σ排斥作用共同影响CO-metal的相互作用。在2014年[21],Nicholas在π-σ模型的基础上,通过分析吸附的CO的电荷、极性、电子密度去判断CO-metal的相互作用。本文借鉴当前的π-σ作用,使用COHP分析和DOS相结合的方式,直观判断各轨道成键强度的变化,从而得出吸附的CO与金属Co的轨道相互作用对CO活化的影响,期待相关研究结果能为CO活化机理的阐明提供一定的有用线索。

Fig.1 Orbital interaction for CO adsorption on Co surface sites图1 CO吸附在Co金属表面之后的轨道相互作用
1 计算模型和方法
1.1 计算模型
钴晶体有两种常见的堆积模型,面心立方结构(Face-Centered Cubic Structure,FCC)和六方最密堆积结构(Hexagonal Close-Packed Structure,HCP)。目前,有很多成果证明HCP Co的活性高于FCC Co[22]。本文以HCP Co为基础进行研究,对体相进行优化,并计算常见表面的表面能。在点群为6/mmm的HCP 钴晶体上,依据表面能数值可产生包含以下几个表面(0001)、(10-11)、(10-10)、(10-14)、(11-21)的Wulff结构图,如图2a所示。根据各表面包含的位点确定研究表面,表面(0001)包含top位,bridge位,fcc位,hcp位四种位点,计算证明hcp位是最容易吸附的位点,本文考虑hcp位点;表面(10-11)含有top位,bridge位,因活性低于hcp位,本文不做考虑;(10-10)表面含两种位点B5和B6(Bn表示位点,n代表吸附位点处与吸附物紧密相连的金属原子的个数),下文分别用(10-10)a和(10-10)b来代表。(10-14)表面含有两种B5位点,原子数目排列分别为2-1-2和2-2-1,下文分别使用(10-14)a和(10-14)b来代表,表面结构如图2b所示,黄色原子代表吸附位点。因而,本文主要研究以下六个表面(0001)、(10-10)a、(10-10)b、(10-14)a、(10-14)b、(11-21)。
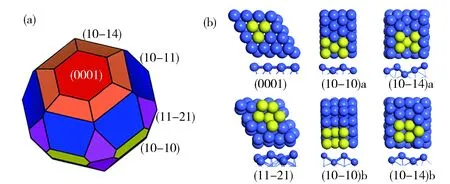
Fig.2 (a) Wulff constructure of a Co hcp nanoparticle;(b)six kinds of surface terminations图2 (a)钴纳米颗粒的Wulff结构图(b)六种钴金属表面
1.2 计算方法
所有的计算均在VASP程序包(Vienna Ab-inito Simulation Package,VASP)完成。电子间的交换相关能计算采用PBE泛函。布里渊区内的积分采用Monkhorst-Pack方案产生不可约K点网格。结构优化中,截断能是400 eV,原子平衡位置的搜索使用的是共轭梯度(CG)算法,总能收敛性判据为10-5eV/atom,原子受力的收敛判据为3×10-2eV/Å,同时考虑了自旋极化的影响。自洽计算的收敛标准是能量达到10-6eV/atom,原子受力小于2×10-2eV/Å。采用Cl-NEB(The Climbing Image Nudge Elastic Band)[23]方法寻找过渡态,弛豫原子沿反应坐标切线方向上的力低于3×10-2eV/Å。
电子结构分析使用晶体轨道哈密顿布居分析(crystal orbital hamilton population analysis,COHP)[24]与态密度分析(density of states,DOS)相结合的方式。COHP 分析可以呈现详细的分子轨道信息,描述体系的成键,非键和反键特征。由于我们使用的是以一组平面波为基组的投影缀加波方法(PAW),故会得到投影COHP曲线(projected COHP,pCOHP)。本文中pCOHP的呈现以其负值-(pCOHP)来表示,相应的正值和负值分别对应成键和反键作用,而数值的绝对值大小表示成键强度。通过投影态密度(projected density of states,pDOS)可以帮助分析各个轨道的贡献。
2 结果与讨论
2.1 热力学分析:CO的吸附和活化
如图3a所示,CO的吸附能对Co的表面结构具有一定的结构敏感性,不同表面上吸附能差别不大,这与Ge等人对CO在各种Co金属表面的吸附能研究结果一致[25]。CO、CHO和COH在不同表面吸附后的C-O键长情况如图3b所示。CO直接吸附的结果用绿色表示。与自由CO的C-O键长1.14 Å相比,CO吸附在(0001)、(10-10)a、(10-14)a表面上时,碳氧键长仅轻微拉长到1.20Å左右;而在表面(10-14)b、(11-21)、(10-10)b上吸附时,C-O键长被显著拉长,最长达1.34 Å。当在碳或氧上引入氢之后,在(0001)、(10-10)a、(10-14)a表面吸附时,C-O键长被显著拉长,吸附在(10-14)b、(11-21)、(10-10)b表面上时,键长也有所变长,其中(11-21)和(10-10)b表面上的-COH物种具有最长的C-O键长。C-O键长的拉长有利于碳氧键的断裂。图3c显示三种吸附物的解离活化能,数值基本上与C-O键长呈反向变化,键长最长的(11-21)和(10-10)b表面上的-COH物种是解离活化能最低的。这说明在我们研究的表面中,CO最容易在H的协助下在(11-21)和(10-10)b表面上发生解离。
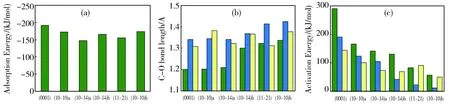
Green:sur-CO;blue:sur-COH; yellow:sur-CHOFig.3 (a) CO adsorption energy; (b) bond distances between carbon and oxygen (c) Activation energy of C-O dissociation. 注:不同颜色代表不同的吸附方式,绿色:CO吸附物种;蓝色:COH吸附物种;黄色:CHO吸附物种图3 (a)CO在不同表面的吸附能;(b)碳氧键长;(c)C-O解离的活化能
2.2 电子结构分析
为了从电子结构的角度对CO的解离机理进行解析,我们对钴与CO之间的轨道相互作用进行了详细的分析:包括CO吸附前的轨道、以(0001)和(10-10)b表面为代表的吸附后的轨道,以及(0001)表面上直接解离和氢助解离两种活化方式下的轨道。
2.2.1 孤立CO分子
孤立CO分子的 pDOS图见图4a。由图可知,CO的主要的占据轨道有3σ、4σ、1π和5σ,而主要的非占据轨道为2π*。对照CO的pCOHP图(图4b)可知,3σ和1π是主要的成键轨道,而4σ和5σ在成键上的贡献较弱。
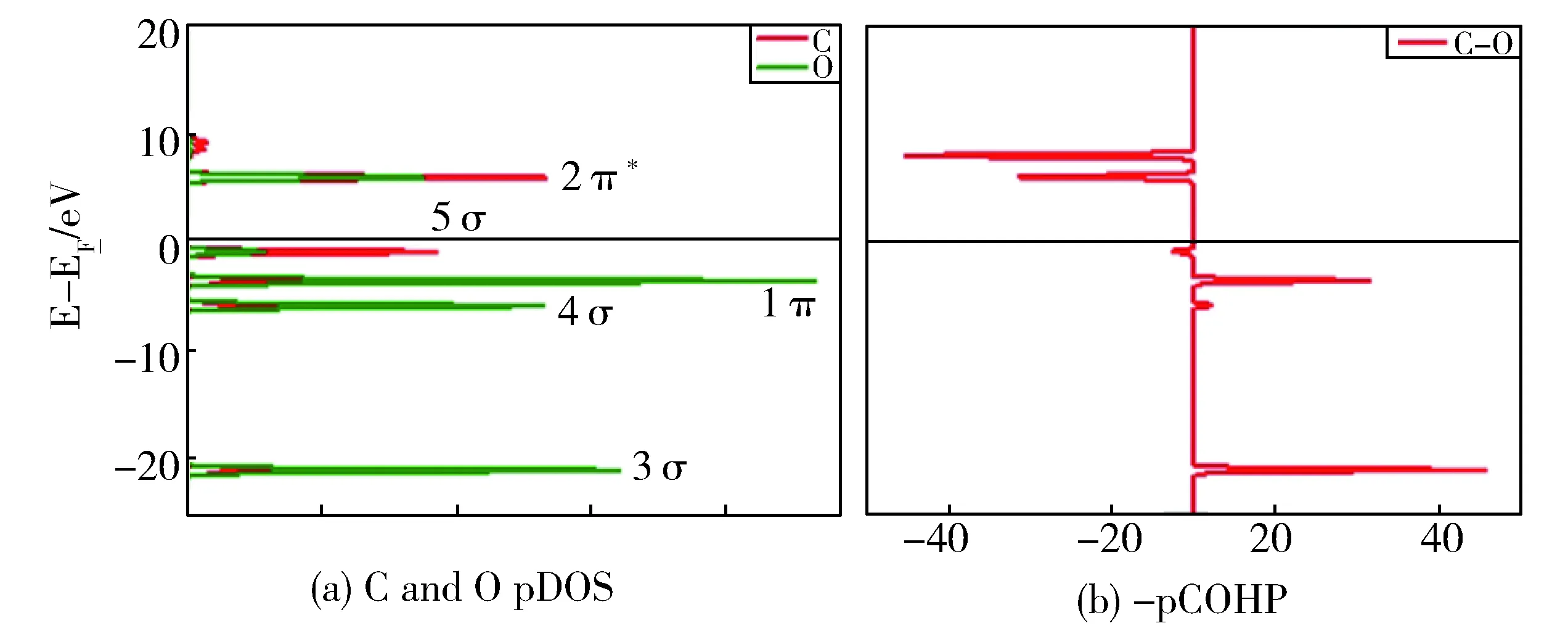
Fig.4 (a) pDOS curve and (b) pCOHP curve for free CO图4 (a)CO分子的投影态密度分析;(b)CO分子的晶体轨道哈密顿分布分析
2.2.2 CO吸附到表面(0001)和(10-10)b
当CO吸附到金属表面上后,由于金属dπ轨道与CO分子的1π和2π*轨道形状对称,能量相近,轨道间发生杂化,形成新的轨道。这种相互作用可以通过CO吸附前后pDOS和pCOHP图的变化得到验证。如图5a所示,CO吸附在(0001)表面上之后,其pDOS图的费米能级附近出现了一个新的dπ杂化轨道(图中对应的杂化轨道以含波浪线的轨道表示),其电子主要来自于氧原子。对照pCOHP图(图5b)可知,在CO分子内,4σ、5σ和1π杂化轨道对C-O起成键作用(红线),而dπ与2π*杂化轨道起反键作用;另外,Co-C的成键相互作用(蓝线)相对较强,因为除了2π*杂化轨道,其他都是成键作用;而Co-O的相互作用(绿线)强度很弱。对比自由的CO分子,Co-CO的相互作用形成了新的轨道,它们即可以明显降低4σ、5σ和1π杂化轨道的能量,稳定吸附体系,又能增加Co-C成键作用而削弱C-O成键强度,从而有利于活化碳氧键。
CO吸附在表面(10-10)b上的pDOS和pCOHP分析结果如图5c和5d所示。从pDOS图可以看出,吸附后新出现的dπ杂化轨道上的电子同时来自于碳和氧,这与吸附在(0001)表面上时电子只来自于氧有明显区别。而pCOHP的分析结果表明,Co除了与碳有显著相互作用(蓝线)外,与氧也有不可忽略的成键作用(绿线),这使得C-O键的强度被进一步削弱,有利于C-O键的活化。同时Co-CO相互作用也使得5σ比1π更稳定,从而整个体系更加稳定。这些结果与吸附能和活化能得到的结果相一致。

Fig.5 (a) pDOS curve and (b)pCOHP curve for CO adsorption on surface (0001)(c) pDOS curve and (d)pCOHP curve for CO adsorption on surface (10-10)b图5 (a) CO分子吸附在表面(0001)上的投影态密度分析; (b) 晶体轨道哈密顿布居分析;(c) CO分子吸附在表面(10-10)b上的投影态密度分析; (d) 晶体轨道哈密顿布居分析。

Fig.6 (a) pDOS curve and (b) pCOHP curve for CO adsorption on surface (0001) in the way of CO-H (c) pDOS curve and (d) pCOHPcurve for CO adsorption on surface (0001) in the way of H-CO图6 (a) CO分子以CO-H吸附物在表面(0001)上的投影态密度分析;(b) 晶体轨道哈密顿布居分析; (c) CO分子以H-CO吸附物在表面(0001)上的投影态密度分析;(d) 晶体轨道哈密顿布居分析
2.2.3 两种机理的对比
在氢原子的协助下,CO与金属的轨道相互作用又会对CO的活化产生什么影响呢?我们以(0001)面为例对两种氢助解离方式的电子结构进行了分析(图6a-6d)。当表面物种是CO-H时,dπ杂化轨道上的电子明显增强碳的参与,电子分布于碳和氧上。当表面物种是H-CO时,dπ杂化轨道上的电子主要来自于氧,碳和氢的成分很少。对比pCOHP曲线(图5b与图6b,6d):氢助之后,C-O成键强度明显减弱,但是表面物种是H-CO时,减弱程度更大,相应活化能更低。另外也可以发现,以H-CO物种吸附解离时,除了C-O和Co-C的成键相互作用外,Co-O的成键作用也被明显增强,同样也协助降低活化能,反应更有利。
3 结论
从电子结构分析的角度对CO在hcp晶型金属Co表面解离的机理进行了详细的分析,得出以下结论:
(1)表面结构对CO的解离有显著影响的电子结构原因:一方面,表面与CO的相互作用会形成dπ杂化轨道,该轨道参与杂化原子所提供电子占的份额越多,CO键的活化程度就越高;另一方面,Co-C键和Co-O键的成键强度越大,CO的吸附就越稳定,越有利于碳氧键活化。
(2)H助机理优于碳化机理的电子结构原因:H连接在O原子上时,H的引入能增加dπ杂化轨道中C的成分;而H连接在C原子上时,H的引入能显著增强Co-O键,增强了吸附体系的稳定性,二者都能显著增加C-O键的活化程度。这些结果表明,通过增加dπ杂化轨道中参与原子的电子份额或加强碳和氧与Co的成键强度都能有助于碳-氧键的活化。
猜你喜欢
无机化学学报(2022年9期)2022-09-16
建材发展导向(2021年14期)2021-08-23
青岛大学学报(工程技术版)(2019年2期)2019-09-10
中国煤层气(2019年2期)2019-08-27
中国塑料(2016年1期)2016-05-17
读写算·教研版(2016年8期)2016-05-07
中国塑料(2016年11期)2016-04-16
枣庄学院学报(2015年5期)2016-01-09
中学化学(2015年8期)2015-12-29
火炸药学报(2014年1期)2014-03-20
